This information is produced and provided by the National Cancer Institute (NCI). The information in this topic may have changed since it was written. For the most current information, contact the National Cancer Institute via the Internet web site at http://cancer.gov or call 1-800-4-CANCER.
General Information About Chronic Lymphocytic Leukemia
Incidence and Mortality
Estimated new cases and deaths from chronic lymphocytic leukemia (CLL) in the United States in 2025:[1]
New cases: 23,690.
Deaths: 4,460.
Anatomy
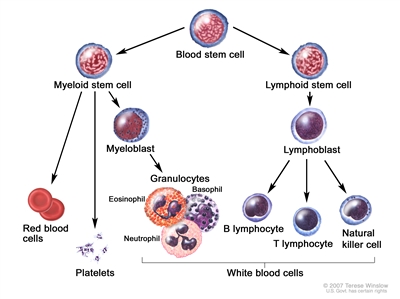
CLL is a disorder of morphologically mature, but immunologically less mature lymphocytes. It is manifested by progressive accumulation of these cells in the blood, bone marrow, and lymphatic tissues.[2]
Blood cell development. A blood stem cell goes through several steps to become a red blood cell, platelet, or white blood cell.
Clinical Presentation
The clinical course of this disease progresses from an indolent lymphocytosis without other evident disease to one of generalized lymphatic enlargement with concomitant pancytopenia. Complications of pancytopenia, including hemorrhage and infection, represent a major cause of death in these patients.[3] Immunological aberrations, including Coombs-positive hemolytic anemia, immune thrombocytopenia, and depressed immunoglobulin levels, may all complicate the management of CLL.[4]
Diagnostic Evaluation and Differential Diagnosis
Tests and procedures used to diagnose CLL include the following:[5]
History and physical examination (including bidimensional diameters of the largest palpable lymph nodes in the cervical, axillary, and inguinal nodal sites and dimensions of the liver and spleen below their respective costal margins as assessed by palpation).
Complete blood count with differential and chemistry panel (including creatinine, bilirubin, transaminases, and alkaline phosphatase). Other blood tests may include lactate dehydrogenase and beta-2-microglobulin. With suspicion of autoimmune hemolytic anemia, testing for reticulocyte count, indirect bilirubin, serum haptoglobin, antiglobulin (direct Coombs), and cold agglutinin may be helpful.
Flow cytometry (for immunophenotyping).
Fluorescence in situ hybridization (FISH) (for del(11q), del(13q), del(17p), trisomy 12, and t(11;14)).
TP53 variant analysis.
IGH variant analysis.
Serum immunoglobulin levels.
Hepatitis B and C and HIV tests.
Computed tomography (CT) is usually not required in the absence of peripheral adenopathy; extensive adenopathy on examination should prompt investigation of retroperitoneal adenopathy.
Bone marrow aspiration and biopsy is usually not required.
In this disorder, lymphocyte counts in the blood are usually greater than or equal to 5,000/mm3 with a characteristic immunophenotype (CD5- and CD23-positive B cells).[6,7] As assays have become more sensitive for detecting monoclonal B-CLL–like cells in peripheral blood, researchers have detected a monoclonal B-cell lymphocytosis in 3% of adults older than 40 years and in 6% of adults older than 60 years.[8] Such early detection and diagnosis may falsely suggest improved survival for the group and may unnecessarily worry or result in therapy for some patients who would have remained undiagnosed in their lifetime, a circumstance known as overdiagnosis or pseudodisease.[9,10]
Confusion with other diseases may be avoided by determination of cell surface markers. CLL lymphocytes coexpress the B-cell antigens CD19 and CD20 along with the T-cell antigen CD5.[11] This coexpression occurs in only one other disease entity, mantle cell lymphoma. CLL B cells express relatively low levels of surface-membrane immunoglobulin (compared with normal peripheral blood B cells) and a single light chain (kappa or lambda).[12] CLL is diagnosed by an absolute increase in lymphocytosis and/or bone marrow infiltration coupled with the characteristic features of morphology and immunophenotype, which confirm the characteristic clonal population. In a database analysis, for up to 77 months before diagnosis, almost all patients with a CLL diagnosis had prediagnostic B-cell clones that were identified in peripheral blood (when available).[7,13]
About 1% of morphological CLL cases express T-cell markers (CD4 and CD7) and have clonal rearrangements of their T-cell receptor genes. These patients have a higher frequency of skin lesions, more variable lymphocyte shape, and shorter median survival (13 months) with minimal responses to chemotherapy and B-cell receptor inhibitors.[14]
The differential diagnosis must exclude the following:
Monoclonal B-cell lymphocytosis (MBL). MBL, the precursor to CLL, is defined as a clonal B-cell population circulating in peripheral blood with fewer than 5 × 109 /L B cells and no signs of lymphadenopathy or splenomegaly.[15] Most cases have the immunophenotype of CLL. The incidence of MBL in the general population is 5% to 12% and increases with age.[16] In families with two or more cases of CLL, MBL has a prevalence of 13% to 18%. Low-count MBL (≤0.5 × 109 /L B cells) rarely progresses to overt CLL, but higher levels can progress to symptomatic CLL at a rate of less than 2% per year, even for familial cases.[15,17] In two selected series of more than 900 patients monitored prospectively for a median of 5 to 7 years, overt CLL requiring chemotherapy occurred in 7% of patients.[8,18] A screening study using the Mayo Clinic Biobank identified 1,712 patients with MBL from 10,139 screened samples.[19] Low-count MBL was found in 95% of these patients. With a median follow-up of 10.0 years, only 0.58% of patients progressed to a lymphoid malignancy.[19]
Waldenström macroglobulinemia. Waldenström macroglobulinemia has a natural history and therapeutic options similar to CLL, with the exception of hyperviscosity syndrome associated with macroglobulinemia as a result of elevated immunoglobulin M. For more information, see Indolent B-Cell Non-Hodgkin Lymphoma Treatment.
Large granular lymphocyte (LGL) leukemia. LGL leukemia is characterized by lymphocytosis with a natural killer (NK) cell immunophenotype (CD2, CD16, and CD56) or a T-cell immunophenotype (CD2, CD3, and CD8).[20,21,22] These patients often have neutropenia and a history of rheumatoid arthritis. The natural history is indolent, often marked by anemia and splenomegaly. This condition appears to fit into the clinical spectrum of Felty syndrome.[23] A characteristic genetic finding in almost 50% of the patients with T-cell LGL involves pathogenic variants in the STAT3 gene.[24] Symptomatic patients with cytopenias typically manifest CD8-positive T cells with alpha/beta surface receptors plus a STAT3 variant, CD8-positive T cells with T gamma/delta surface receptors, or a mutated NK/T-cell phenotype.[25,26] Conversely, asymptomatic patients have wild-type STAT3 CD8-positive T cells, CD4-positive T cells, and wild-type NK cells.[25] Therapy includes low doses of oral cyclophosphamide or methotrexate, cyclosporine, and treatment of the bacterial infections acquired during severe neutropenia.[20,22,27,28]
For information on prolymphocytic leukemia, which was previously covered in this summary, see the Treatment of T-Cell Prolymphocytic Leukemia section in Peripheral T-Cell Non-Hodgkin Lymphoma Treatment.
Prognostic Factors
Prognostic markers help stratify patients in clinical trials, assess the need for therapy, and select the type of therapy.[2,29,30] Prognostic factors that may help predict clinical outcome include cytogenetic subgroup, immunoglobulin mutational status, and CD38 immunophenotype.[2,31,32,33,34,35,36,37,38,39]
Prognostic markers include the following:
IGH pathogenic variant.[32,33,34,39,40] The finding of significant numbers of variants in this region is associated with a median survival in excess of 20 to 25 years. The absence of variants is associated with a median survival of 8 to 10 years.
FISH test results. FISH chromosomal abnormalities were associated with prognosis in retrospective and prospective studies, and clonal evolution has been seen over time.[31,41,42,43] The following chromosomal abnormalities have been reported:
del(13q) is a favorable prognostic marker (median overall survival [OS], 17 years in one prospective study).[43]
Trisomy 12 and del(11) have a less favorable prognosis (median OS, 9–11 years in one prospective study).[43]
del(17p) is associated with TP53 pathogenic variants, poor response rates, and short duration of response to the standard therapeutic options.[39] del(17p) is associated with the most unfavorable prognosis (median OS, 7 years in one prospective trial).[43,44,45]
The combination of abnormal cytogenetics, such as del(11q) or del(17p) deletion (suggesting a worse prognosis), with zeta-chain-associated protein 70 kDa negativity (suggesting a better prognosis) in the same patients resulted in a poor prognosis.[38]
These findings emphasize the need for prospective studies of combinations of these prognostic markers.[46]
Other prognostic factors include the following:
Anemia and thrombocytopenia. These are important adverse prognostic variables, but only if due to extensive marrow involvement by CLL. Autoimmune hemolytic anemia and immune thrombocytopenic purpura do not confer a worse prognosis.
Age. CLL occurs primarily in middle-aged and older adults, with worse prognosis in successive decades of life.[42]
Positron emission tomography (PET)-CT scan results. This test should only be used in the context of recurrent fever, soaking night sweats, weight loss (>10% baseline weight in 6 months), or rapidly growing lymph nodes, because these findings might herald histological transformation to a diffuse large B-cell lymphoma (DLBCL) (so-called Richter transformation). Of 432 patients retrospectively reviewed, 209 patients had a maximum standardized uptake value (SUVmax) of 5 or higher.[49] Eighty percent of these patients had histologically aggressive CLL or Richter syndrome, and both of these entities had equally worse prognoses. When the SUVmax was 10 or higher, the 5-year OS rate was only 30%.[49]
Lymphocyte doubling time. Doubling of the white blood cell count in less than 1 year implies a worse prognosis.[50]
Beta-2-microglobulin. Higher levels imply a worse prognosis.[51]
Richter transformation. In 2% to 10% of patients, CLL will transform into a more aggressive lymphoma, termed Richter transformation.[52] This is usually a DLBCL of the more aggressive activated B-cell subtype. The prognosis is similar to de novo presentations of DLBCL when the CLL has never required therapy or when there is no clonal connection between the CLL and DLBCL (identified if they harbor different clonal light chains or if sequencing can be accomplished at the VDJ [variable, diversity, joining] recombination sites in the immunoglobulin heavy chain variable region).[52] However, the prognosis is poor (median survival, 6–14 months) for most patients with Richter transformation to DLBCL when there has been prior therapy for CLL with chemoimmunotherapy,[53], Bruton tyrosine kinase (BTK) inhibitors, and/or venetoclax.[54] There is no standard therapeutic approach for patients with poor prognoses. Therapies under clinical evaluation include chimeric antigen receptor (CAR) T-cell therapy, T-cell engaging bispecific antibodies, and non-covalent BTK inhibitors.[52,55] Allogeneic stem cell transplant consolidation is often recommended if induction therapy achieves a response.[52] In rare cases of Richter transformation from CLL to Hodgkin lymphoma, the prognosis appears the same for age-matched patients with de novo disease.[56,57]
Clearance of measurable residual disease (MRD). The improvements in response rates from more intensive regimens have maximized the clearance of MRD. In one prospective trial of 493 patients, clearance of MRD was an independent predictor of OS by multivariate analysis.[58] The surrogate end point of clearance of residual disease, while prognostic,[58,59] did not show improved survival in a randomized prospective trial. The necessary study would include patients who fail to completely clear the marrow with induction therapy and randomly assign them to further alternative treatment versus the same treatment later at relapse, looking at OS as the primary end point.[29,60]
CD38 immunophenotype.[33,61] CD38 positivity (>30%) correlates with a worse prognosis, but there is a 30% false-positive rate and a 50% false-negative rate using IGH mutational status as the gold standard for prognosis.
Other malignancies. Patients with CLL are also at increased risk of developing other malignancies, even before therapy.[62] A population-based analysis of almost 2 million cancer patients in the National Cancer Institute's Surveillance, Epidemiology, and End Results (SEER) Program database was performed. The findings suggested that cancer-specific survival for patients with preexisting CLL who subsequently developed colorectal and breast cancer was significantly lower (hazard ratio [HR], 1.46; P < .001 for colorectal cancer and HR, 1.41; P = .005 for breast cancer) than cancer-specific survival for patients with colorectal and breast cancer who did not have antecedent CLL, after adjusting for age, sex, race, and disease stage, and excluding CLL-related deaths.[63]
An international prognostic index for CLL (CLL-IPI) identified four prognostic subgroups based on IGH mutational status, clinical stage, age (≤65 years vs. >65 years), and TP53 status (no abnormalities vs. del(17p), TP53 variant, or both).[64] A scoring system to predict time to first treatment for early-stage CLL identified three adverse risk factors: unmutated IGH, absolute lymphocyte count higher than 15 × 109 /L, and palpable lymph nodes.[65] Any new prognostic model, and even the commonly used CLL-IPI, may be outdated because of the use of highly effective frontline therapies, including BCL2 inhibitors and BTK inhibitors.[66] Revalidation of these prognostic models will be required.
Follow-Up After Treatment
CT scans have a very limited role in monitoring patients after completion of treatment. CT scan or ultrasonography results determined the decision to treat for relapse in only 2 of 176 patients in three prospective trials for the German CLL Study Group.[67]
References:
American Cancer Society: Cancer Facts and Figures 2025. American Cancer Society, 2025. Available online. Last accessed January 16, 2025.
Burger JA: Treatment of Chronic Lymphocytic Leukemia. N Engl J Med 383 (5): 460-473, 2020.
Anaissie EJ, Kontoyiannis DP, O'Brien S, et al.: Infections in patients with chronic lymphocytic leukemia treated with fludarabine. Ann Intern Med 129 (7): 559-66, 1998.
Mauro FR, Foa R, Cerretti R, et al.: Autoimmune hemolytic anemia in chronic lymphocytic leukemia: clinical, therapeutic, and prognostic features. Blood 95 (9): 2786-92, 2000.
Hallek M, Cheson BD, Catovsky D, et al.: iwCLL guidelines for diagnosis, indications for treatment, response assessment, and supportive management of CLL. Blood 131 (25): 2745-2760, 2018.
Hallek M, Cheson BD, Catovsky D, et al.: Guidelines for the diagnosis and treatment of chronic lymphocytic leukemia: a report from the International Workshop on Chronic Lymphocytic Leukemia updating the National Cancer Institute-Working Group 1996 guidelines. Blood 111 (12): 5446-56, 2008.
Shanafelt TD, Kay NE, Jenkins G, et al.: B-cell count and survival: differentiating chronic lymphocytic leukemia from monoclonal B-cell lymphocytosis based on clinical outcome. Blood 113 (18): 4188-96, 2009.
Rawstron AC, Bennett FL, O'Connor SJ, et al.: Monoclonal B-cell lymphocytosis and chronic lymphocytic leukemia. N Engl J Med 359 (6): 575-83, 2008.
Dighiero G: Monoclonal B-cell lymphocytosis--a frequent premalignant condition. N Engl J Med 359 (6): 638-40, 2008.
Fazi C, Scarfò L, Pecciarini L, et al.: General population low-count CLL-like MBL persists over time without clinical progression, although carrying the same cytogenetic abnormalities of CLL. Blood 118 (25): 6618-25, 2011.
DiGiuseppe JA, Borowitz MJ: Clinical utility of flow cytometry in the chronic lymphoid leukemias. Semin Oncol 25 (1): 6-10, 1998.
Rozman C, Montserrat E: Chronic lymphocytic leukemia. N Engl J Med 333 (16): 1052-7, 1995.
Landgren O, Albitar M, Ma W, et al.: B-cell clones as early markers for chronic lymphocytic leukemia. N Engl J Med 360 (7): 659-67, 2009.
Hoyer JD, Ross CW, Li CY, et al.: True T-cell chronic lymphocytic leukemia: a morphologic and immunophenotypic study of 25 cases. Blood 86 (3): 1163-9, 1995.
Shim YK, Rachel JM, Ghia P, et al.: Monoclonal B-cell lymphocytosis in healthy blood donors: an unexpectedly common finding. Blood 123 (9): 1319-26, 2014.
Slager SL, Lanasa MC, Marti GE, et al.: Natural history of monoclonal B-cell lymphocytosis among relatives in CLL families. Blood 137 (15): 2046-2056, 2021.
Shanafelt TD, Kay NE, Rabe KG, et al.: Brief report: natural history of individuals with clinically recognized monoclonal B-cell lymphocytosis compared with patients with Rai 0 chronic lymphocytic leukemia. J Clin Oncol 27 (24): 3959-63, 2009.
Slager SL, Parikh SA, Achenbach SJ, et al.: Progression and survival of MBL: a screening study of 10 139 individuals. Blood 140 (15): 1702-1709, 2022.
Semenzato G, Zambello R, Starkebaum G, et al.: The lymphoproliferative disease of granular lymphocytes: updated criteria for diagnosis. Blood 89 (1): 256-60, 1997.
Lamy T, Loughran TP: How I treat LGL leukemia. Blood 117 (10): 2764-74, 2011.
Bowman SJ, Sivakumaran M, Snowden N, et al.: The large granular lymphocyte syndrome with rheumatoid arthritis. Immunogenetic evidence for a broader definition of Felty's syndrome. Arthritis Rheum 37 (9): 1326-30, 1994.
Koskela HL, Eldfors S, Ellonen P, et al.: Somatic STAT3 mutations in large granular lymphocytic leukemia. N Engl J Med 366 (20): 1905-13, 2012.
Cheon H, Xing JC, Moosic KB, et al.: Genomic landscape of TCRαβ and TCRγδ T-large granular lymphocyte leukemia. Blood 139 (20): 3058-3072, 2022.
Barilà G, Grassi A, Cheon H, et al.: Tγδ LGLL identifies a subset with more symptomatic disease: analysis of an international cohort of 137 patients. Blood 141 (9): 1036-1046, 2023.
Loughran TP, Kidd PG, Starkebaum G: Treatment of large granular lymphocyte leukemia with oral low-dose methotrexate. Blood 84 (7): 2164-70, 1994.
Dhodapkar MV, Li CY, Lust JA, et al.: Clinical spectrum of clonal proliferations of T-large granular lymphocytes: a T-cell clonopathy of undetermined significance? Blood 84 (5): 1620-7, 1994.
Developments in the treatment of lymphoproliferative disorders: rising to the new challenges of CLL therapy. A report of a symposium presented during the 48th American Society of Hematology Annual Meeting and Exposition, December 8, 2006, Orlando, Florida. Clin Adv Hematol Oncol 5 (3 Suppl 5): 1-14; quiz 15-6, 2007.
Pflug N, Bahlo J, Shanafelt TD, et al.: Development of a comprehensive prognostic index for patients with chronic lymphocytic leukemia. Blood 124 (1): 49-62, 2014.
Döhner H, Stilgenbauer S, Benner A, et al.: Genomic aberrations and survival in chronic lymphocytic leukemia. N Engl J Med 343 (26): 1910-6, 2000.
Hamblin TJ, Davis Z, Gardiner A, et al.: Unmutated Ig V(H) genes are associated with a more aggressive form of chronic lymphocytic leukemia. Blood 94 (6): 1848-54, 1999.
Damle RN, Wasil T, Fais F, et al.: Ig V gene mutation status and CD38 expression as novel prognostic indicators in chronic lymphocytic leukemia. Blood 94 (6): 1840-7, 1999.
Rosenwald A, Alizadeh AA, Widhopf G, et al.: Relation of gene expression phenotype to immunoglobulin mutation genotype in B cell chronic lymphocytic leukemia. J Exp Med 194 (11): 1639-47, 2001.
Klein U, Tu Y, Stolovitzky GA, et al.: Gene expression profiling of B cell chronic lymphocytic leukemia reveals a homogeneous phenotype related to memory B cells. J Exp Med 194 (11): 1625-38, 2001.
Orchard JA, Ibbotson RE, Davis Z, et al.: ZAP-70 expression and prognosis in chronic lymphocytic leukaemia. Lancet 363 (9403): 105-11, 2004.
Rassenti LZ, Huynh L, Toy TL, et al.: ZAP-70 compared with immunoglobulin heavy-chain gene mutation status as a predictor of disease progression in chronic lymphocytic leukemia. N Engl J Med 351 (9): 893-901, 2004.
Kröber A, Bloehdorn J, Hafner S, et al.: Additional genetic high-risk features such as 11q deletion, 17p deletion, and V3-21 usage characterize discordance of ZAP-70 and VH mutation status in chronic lymphocytic leukemia. J Clin Oncol 24 (6): 969-75, 2006.
Byrd JC, Gribben JG, Peterson BL, et al.: Select high-risk genetic features predict earlier progression following chemoimmunotherapy with fludarabine and rituximab in chronic lymphocytic leukemia: justification for risk-adapted therapy. J Clin Oncol 24 (3): 437-43, 2006.
Kharfan-Dabaja MA, Chavez JC, Khorfan KA, et al.: Clinical and therapeutic implications of the mutational status of IgVH in patients with chronic lymphocytic leukemia. Cancer 113 (5): 897-906, 2008.
Kröber A, Seiler T, Benner A, et al.: V(H) mutation status, CD38 expression level, genomic aberrations, and survival in chronic lymphocytic leukemia. Blood 100 (4): 1410-6, 2002.
Catovsky D, Fooks J, Richards S: Prognostic factors in chronic lymphocytic leukaemia: the importance of age, sex and response to treatment in survival. A report from the MRC CLL 1 trial. MRC Working Party on Leukaemia in Adults. Br J Haematol 72 (2): 141-9, 1989.
Shanafelt TD, Witzig TE, Fink SR, et al.: Prospective evaluation of clonal evolution during long-term follow-up of patients with untreated early-stage chronic lymphocytic leukemia. J Clin Oncol 24 (28): 4634-41, 2006.
Grever MR, Lucas DM, Dewald GW, et al.: Comprehensive assessment of genetic and molecular features predicting outcome in patients with chronic lymphocytic leukemia: results from the US Intergroup Phase III Trial E2997. J Clin Oncol 25 (7): 799-804, 2007.
Catovsky D, Richards S, Matutes E, et al.: Assessment of fludarabine plus cyclophosphamide for patients with chronic lymphocytic leukaemia (the LRF CLL4 Trial): a randomised controlled trial. Lancet 370 (9583): 230-9, 2007.
Binet JL, Caligaris-Cappio F, Catovsky D, et al.: Perspectives on the use of new diagnostic tools in the treatment of chronic lymphocytic leukemia. Blood 107 (3): 859-61, 2006.
Rai KR, Sawitsky A, Cronkite EP, et al.: Clinical staging of chronic lymphocytic leukemia. Blood 46 (2): 219-34, 1975.
Binet JL, Auquier A, Dighiero G, et al.: A new prognostic classification of chronic lymphocytic leukemia derived from a multivariate survival analysis. Cancer 48 (1): 198-206, 1981.
Falchi L, Keating MJ, Marom EM, et al.: Correlation between FDG/PET, histology, characteristics, and survival in 332 patients with chronic lymphoid leukemia. Blood 123 (18): 2783-90, 2014.
Montserrat E, Sanchez-Bisono J, Viñolas N, et al.: Lymphocyte doubling time in chronic lymphocytic leukaemia: analysis of its prognostic significance. Br J Haematol 62 (3): 567-75, 1986.
Di Giovanni S, Valentini G, Carducci P, et al.: Beta-2-microglobulin is a reliable tumor marker in chronic lymphocytic leukemia. Acta Haematol 81 (4): 181-5, 1989.
Parikh SA, Kay NE, Shanafelt TD: How we treat Richter syndrome. Blood 123 (11): 1647-57, 2014.
Rossi D, Spina V, Deambrogi C, et al.: The genetics of Richter syndrome reveals disease heterogeneity and predicts survival after transformation. Blood 117 (12): 3391-401, 2011.
Kittai AS, Huang Y, Miller S, et al.: Outcomes of patients with Richter transformation without prior chemoimmunotherapy for CLL/SLL: an international multicenter retrospective study. [Abstract] Blood 142 (Suppl 1): A-497, 2023.
Kittai AS, Bond D, Huang Y, et al.: Anti-CD19 Chimeric Antigen Receptor T-Cell Therapy for Richter Transformation: An International, Multicenter, Retrospective Study. J Clin Oncol 42 (17): 2071-2079, 2024.
Al-Sawaf O, Robrecht S, Bahlo J, et al.: Richter transformation in chronic lymphocytic leukemia (CLL)-a pooled analysis of German CLL Study Group (GCLLSG) front line treatment trials. Leukemia 35 (1): 169-176, 2021.
Stephens DM, Boucher K, Kander E, et al.: Hodgkin lymphoma arising in patients with chronic lymphocytic leukemia: outcomes from a large multi-center collaboration. Haematologica 106 (11): 2845-2852, 2021.
Böttcher S, Ritgen M, Fischer K, et al.: Minimal residual disease quantification is an independent predictor of progression-free and overall survival in chronic lymphocytic leukemia: a multivariate analysis from the randomized GCLLSG CLL8 trial. J Clin Oncol 30 (9): 980-8, 2012.
Strati P, Keating MJ, O'Brien SM, et al.: Eradication of bone marrow minimal residual disease may prompt early treatment discontinuation in CLL. Blood 123 (24): 3727-32, 2014.
Montserrat E, Moreno C, Esteve J, et al.: How I treat refractory CLL. Blood 107 (4): 1276-83, 2006.
Ghia P, Guida G, Stella S, et al.: The pattern of CD38 expression defines a distinct subset of chronic lymphocytic leukemia (CLL) patients at risk of disease progression. Blood 101 (4): 1262-9, 2003.
Tsimberidou AM, Wen S, McLaughlin P, et al.: Other malignancies in chronic lymphocytic leukemia/small lymphocytic lymphoma. J Clin Oncol 27 (6): 904-10, 2009.
Solomon BM, Rabe KG, Slager SL, et al.: Overall and cancer-specific survival of patients with breast, colon, kidney, and lung cancers with and without chronic lymphocytic leukemia: a SEER population-based study. J Clin Oncol 31 (7): 930-7, 2013.
International CLL-IPI working group: An international prognostic index for patients with chronic lymphocytic leukaemia (CLL-IPI): a meta-analysis of individual patient data. Lancet Oncol 17 (6): 779-90, 2016.
Condoluci A, Terzi di Bergamo L, Langerbeins P, et al.: International prognostic score for asymptomatic early-stage chronic lymphocytic leukemia. Blood 135 (21): 1859-1869, 2020.
Kreuzberger N, Damen JA, Trivella M, et al.: Prognostic models for newly-diagnosed chronic lymphocytic leukaemia in adults: a systematic review and meta-analysis. Cochrane Database Syst Rev 7: CD012022, 2020.
Eichhorst BF, Fischer K, Fink AM, et al.: Limited clinical relevance of imaging techniques in the follow-up of patients with advanced chronic lymphocytic leukemia: results of a meta-analysis. Blood 117 (6): 1817-21, 2011.
Stage Information for Chronic Lymphocytic Leukemia
Chronic lymphocytic leukemia (CLL) does not have a standard staging system. The Rai staging system (Table 1) and the Binet classification (Table 2) are presented below.[1,2] A National Cancer Institute (NCI)-sponsored working group has formulated standardized guidelines for criteria related to eligibility, response, and toxic effects to be used in future clinical trials in CLL.[3]
Rai Staging System
Table 1. Rai Staging System
Stage
Stage Criteria
Stage 0
Absolute lymphocytosis (>15,000/mm3) without adenopathy, hepatosplenomegaly, anemia, or thrombocytopenia.
Stage I
Absolute lymphocytosis with lymphadenopathy without hepatosplenomegaly, anemia, or thrombocytopenia.
Stage II
Absolute lymphocytosis with either hepatomegaly or splenomegaly with or without lymphadenopathy.
Stage III
Absolute lymphocytosis and anemia (hemoglobin <11 g/dL) with or without lymphadenopathy, hepatomegaly, or splenomegaly.
Stage IV
Absolute lymphocytosis and thrombocytopenia (<100,000/mm3) with or without lymphadenopathy, hepatomegaly, splenomegaly, or anemia.
Binet Classification
Table 2. Binet Classification System
Stage
Stage Criteria
a Lymphoid areas include cervical, axillary, inguinal, and splenic.
Clinical stage Aa
No anemia or thrombocytopenia and fewer than three areas of lymphoid involvement (Rai stages 0, I, and II).
Clinical stage Ba
No anemia or thrombocytopenia with three or more areas of lymphoid involvement (Rai stages I and II).
Clinical stage C
Anemia and/or thrombocytopenia regardless of the number of areas of lymphoid enlargement (Rai stages III and IV).
The Binet classification integrates the number of disease-involved nodal groups with bone marrow failure. Its major benefit derives from the recognition of a predominantly splenic form of the disease, which may have a better prognosis than was recognized in the Rai staging, and from the recognition that the presence of anemia or thrombocytopenia has a similar prognosis and does not merit a separate stage. Neither system separates immune from nonimmune causes of cytopenia. Patients with thrombocytopenia, anemia, or both, which is caused by extensive marrow infiltration and impaired production (Rai III/IV, Binet C), have a poorer prognosis than patients with immune cytopenias.[4]
The International Workshop on CLL has recommended integrating the Rai and Binet systems as follows: A(0), A(I), A(II); B(I), B(II); and C(III), C(IV).[5] The NCI-sponsored working group has published guidelines for the diagnosis and treatment of CLL in both clinical trial and general practice settings.[3] Use of these systems allows comparison of clinical results and establishment of therapeutic guidelines.
References:
Rai KR, Sawitsky A, Cronkite EP, et al.: Clinical staging of chronic lymphocytic leukemia. Blood 46 (2): 219-34, 1975.
Binet JL, Auquier A, Dighiero G, et al.: A new prognostic classification of chronic lymphocytic leukemia derived from a multivariate survival analysis. Cancer 48 (1): 198-206, 1981.
Hallek M, Cheson BD, Catovsky D, et al.: Guidelines for the diagnosis and treatment of chronic lymphocytic leukemia: a report from the International Workshop on Chronic Lymphocytic Leukemia updating the National Cancer Institute-Working Group 1996 guidelines. Blood 111 (12): 5446-56, 2008.
Moreno C, Hodgson K, Ferrer G, et al.: Autoimmune cytopenia in chronic lymphocytic leukemia: prevalence, clinical associations, and prognostic significance. Blood 116 (23): 4771-6, 2010.
Chronic lymphocytic leukemia: recommendations for diagnosis, staging, and response criteria. International Workshop on Chronic Lymphocytic Leukemia. Ann Intern Med 110 (3): 236-8, 1989.
Selection of Therapy for Chronic Lymphocytic Leukemia
Treatment of patients with chronic lymphocytic leukemia (CLL) must be individualized based on the clinical behavior of the disease.[1] Because this disease is generally not curable, occurs in an older population, and often progresses slowly, it is most often treated in a conservative fashion.[2]
In older trials with data collected from the 1970s through the 1990s, the median survival for all patients ranged from 8 to 12 years.[3,4] However, with the introduction of the B-cell receptor inhibitors and targeting of BCL2, the median survival for all patients has not been reached with over 10 years of follow-up.
Treatment of patients with CLL ranges from observation with treatment of infectious, hemorrhagic, or immunological complications to a variety of therapeutic options administered as single agents or combination therapy. In asymptomatic patients, treatment may be deferred until the disease progresses and symptoms occur.[3] Because the rate of progression may vary from patient to patient, with long periods of stability and sometimes spontaneous regressions, frequent and careful observation is required to monitor the clinical course.[5] Although even asymptomatic patients with del(17p) on fluorescence in situ hybridization (FISH) analysis (or those with a TP53 pathogenic variant) may be followed with watchful waiting, frequent monitoring may be required to avert rapid progression. A meta-analysis of randomized trials showed no survival benefit for immediate versus delayed therapy for patients with early-stage disease.[6][Level of evidence A1] For patients with progressing CLL, treatment will not be curative in most cases. Selected patients treated with allogeneic stem cell transplant have achieved prolonged disease-free survival (DFS), sometimes exceeding 20 years.[7,8,9,10,11] Prolonged DFS was also noted in young patients (<60 years) with IGH hypermutation who received the FCR regimen (fludarabine, cyclophosphamide, and rituximab).[12,13,14]
The following clinical factors may be helpful in predicting progression of disease:[2]
IGH pathogenic variant.
Chromosomal abnormalities found by FISH or cytogenetic analysis.
Beta-2-microglobulin.
Lymphocyte doubling time.
The International Workshop on Chronic Lymphocytic Leukemia defines symptomatic or progressive CLL as having the following signs and symptoms:[15]
Evidence of progressive marrow failure—the development or worsening of anemia and/or thrombocytopenia (in some patients, platelet counts <100 × 109 /L may remain stable over a long period; this does not automatically require therapeutic intervention). Cutoff levels of hemoglobin less than 10 g/dL or platelet counts less than 50 × 109 /L are generally regarded as an indication for treatment.
Massive (i.e., ≥6 cm below the left costal margin), progressive, or symptomatic splenomegaly.
Massive lymph nodes (i.e., ≥10 cm in longest diameter), progressive, or symptomatic lymphadenopathy.
Progressive lymphocytosis with an increase of 50% or more over a 2-month period, or lymphocyte-doubling time (LDT) less than 6 months. LDT can be obtained by linear regression extrapolation of absolute lymphocyte counts obtained at intervals of 2 weeks over an observation period of 2 to 3 months; patients with initial blood lymphocyte counts less than 30 × 109 /L may require a longer observation period to determine the LDT. Factors contributing to lymphocytosis other than CLL (e.g., infections or steroid administration) should be excluded.
Autoimmune complications, including anemia or thrombocytopenia that respond poorly to corticosteroids.
Symptomatic or functional extranodal involvement (e.g., skin, kidney, lung, or spine). Disease-related symptoms defined as any of the following:
Unintentional weight loss of 10% or more within the previous 6 months.
Significant fatigue (i.e., Eastern Cooperative Oncology Group performance scale 2 or worse, cannot work, or unable to perform usual activities).
Fevers of 100.5°F or 38.0°C or higher for 2 or more weeks without evidence of infection.
Night sweats for at least 1 month without evidence of infection.
Considerations for the Selection of Therapy
The following general principles may provide a sequencing for available therapeutic options:
Despite many therapeutic options, asymptomatic or minimally affected patients with CLL are often offered observation outside the context of a clinical trial. Therapy often begins when patients develop profound cytopenias, or when symptoms, such as enlarging bulky lymphadenopathy or debilitating symptoms, substantially impact their quality of life.
Because nontransplant curative therapy has not been found, the initial goal of therapy is to maximize efficacy (with improvement of overall survival), while introducing the least overall short- and long-term toxicity.
The U.S. Food and Drug Administration approved the biological agents ibrutinib, acalabrutinib, and venetoclax for first-line use in newly diagnosed patients with CLL who require therapy.[16] In patients with poor prognostic factors (especially those with del(17p) or TP53 pathogenic variants), ibrutinib, acalabrutinib, or venetoclax should be considered.[17]
Standard chemotherapeutic agents, such as fludarabine, bendamustine, cyclophosphamide, and chlorambucil, induce DNA damage that can manifest as more aggressive and refractory phenotypes upon relapse and can induce secondary malignancies. Yet, prolonged DFS (over 10 years) can be seen with the use of the FCR regimen in younger patients (<60 years) with IGH hypermutation.[12,13,14]
Avoiding alkylating agents and purine analogues also prevents prolonged cytopenias and the recurrent, long-lasting, and sometimes fatal infections seen after therapy with these agents.
Avoiding chemotherapeutic agents up-front, when possible, is a new paradigm of sequencing therapy for CLL.
Older patients with comorbidities may better tolerate the newer biological agents (such as ibrutinib or venetoclax), monoclonal antibody therapy alone (such as high-dose rituximab), or dose modification of standard chemotherapeutic agents combined with rituximab. For older patients (>65 years), the combination of rituximab plus bendamustine (BR regimen) resulted in fewer adverse events and better outcomes than the FCR regimen.[18]
Adverse Sequelae of the Disease and Therapy
Infectious complications in advanced disease are in part a consequence of the hypogammaglobulinemia and the inability to mount a humoral defense against bacterial or viral agents. Herpes zoster represents a frequent viral infection in these patients, but infections with Pneumocystis carinii and Candida albicans may also occur. The early recognition of infections and the institution of appropriate therapy are critical to the long-term survival of these patients. A randomized study of intravenous immunoglobulin (400 mg/kg every 3 weeks for 1 year) in patients with CLL and hypogammaglobulinemia produced significantly fewer bacterial infections and a significant delay in onset of first infection during the study period.[19] There was, however, no effect on survival. Routine chronic administration of intravenous immunoglobulin is expensive, and the long-term benefit (>1 year) is unproven.[20,21]
Patients with CLL who required hospitalization for COVID-19 prior to the induction of vaccines fared poorly regardless of stage in two retrospective reports.[22,23] One of the studies noted a protective effect from Bruton tyrosine kinase (BTK) inhibitors (usually ibrutinib),[23] but this was not seen in the other report.[22] With enhanced testing and the advent of multiple therapeutic strategies to prevent and treat COVID-19, the case fatality rate for patients with CLL dropped from 35% in early 2020 to 11% in late 2020 and early 2021 (P < .001).[22] For patients requiring hospitalization, the case fatality rate dropped from 40% to 20% (P = .003).
Autoimmune hemolytic anemia and/or thrombocytopenia can occur in patients with any stage of CLL.[24] Initial therapy involves corticosteroids with or without alkylating agents (fludarabine can worsen the hemolytic anemia). It is often necessary to control the autoimmune destruction with corticosteroids, if possible, before administering marrow-suppressive chemotherapy because it may be difficult for a patient to successfully receive a red blood cell or platelet transfusion. Alternate therapies include high-dose immune globulin, rituximab, cyclosporine, azathioprine, splenectomy, and low-dose radiation therapy to the spleen.[3,25] Tumor lysis syndrome is an uncommon complication (presenting in 1 of 300 patients) of chemotherapy for patients with bulky disease.[26]
Second malignancies and treatment-induced acute leukemias may also occur in a small percentage of patients.[27] Transformation of CLL to diffuse large B-cell lymphoma (DLBCL, known as Richter syndrome) occurs in 2% to 10% of patients. Risk factors for transformation include unmutated IGH, TP53 or NOTCH1 pathogenic variants, CDKN2A/B loss, and a complex karyotype.[28] Up to 60% to 70% of patients develop a DLBCL clonally related to the CLL, and these patients have a significantly worse prognosis than patients with de novo DLBCL.[29,30,31] Patients with a clonally unrelated DLBCL have a much better prognosis, which is similar to de novo DLBCL.[29] However, there is limited availability for real-life sequencing of the immunoglobulin heavy chains in the original CLL sample to compare with the transformed sample. Characteristic molecular signatures may serve as an alternate way to assess prognosis.[32] Up to 20% to 40% of patients with clonally related Richter syndrome are disease free for more than 5 years after aggressive combination chemotherapy, typically R-CHOP (rituximab, cyclophosphamide, doxorubicin, vincristine, and prednisone) or Pola-R-CHP (polatuzumab, rituximab, cyclophosphamide, doxorubicin, and prednisone), often followed by autologous or allogeneic stem cell transplant.[33,34,35] For more information, see Aggressive B-Cell Non-Hodgkin Lymphoma Treatment. In two retrospective reports, CLL with transformation to Hodgkin lymphoma had the same prognosis as de novo presentations of Hodgkin lymphoma at an equivalent age.[36,37][Level of evidence C3]
The BTK inhibitors increased the risk of bleeding requiring hospitalization (3-year risk for patients who received ibrutinib, 8.8% [95% confidence interval (CI), 6.5%–11.7%]) and atrial fibrillation (3-year incidence for patients who received ibrutinib, 22.7% [95% CI, 19.0%–26.6%]).[38] A randomized trial with a median follow-up of 41 months showed less atrial fibrillation for patients with CLL who received acalabrutinib compared with ibrutinib (9.4% vs. 16%; P = .02).[39]
Gribben JG, O'Brien S: Update on therapy of chronic lymphocytic leukemia. J Clin Oncol 29 (5): 544-50, 2011.
Rozman C, Montserrat E: Chronic lymphocytic leukemia. N Engl J Med 333 (16): 1052-7, 1995.
Wierda WG, O'Brien S, Wang X, et al.: Prognostic nomogram and index for overall survival in previously untreated patients with chronic lymphocytic leukemia. Blood 109 (11): 4679-85, 2007.
Del Giudice I, Chiaretti S, Tavolaro S, et al.: Spontaneous regression of chronic lymphocytic leukemia: clinical and biologic features of 9 cases. Blood 114 (3): 638-46, 2009.
Chemotherapeutic options in chronic lymphocytic leukemia: a meta-analysis of the randomized trials. CLL Trialists' Collaborative Group. J Natl Cancer Inst 91 (10): 861-8, 1999.
Ritgen M, Stilgenbauer S, von Neuhoff N, et al.: Graft-versus-leukemia activity may overcome therapeutic resistance of chronic lymphocytic leukemia with unmutated immunoglobulin variable heavy-chain gene status: implications of minimal residual disease measurement with quantitative PCR. Blood 104 (8): 2600-2, 2004.
Moreno C, Villamor N, Colomer D, et al.: Allogeneic stem-cell transplantation may overcome the adverse prognosis of unmutated VH gene in patients with chronic lymphocytic leukemia. J Clin Oncol 23 (15): 3433-8, 2005.
Khouri IF, Keating MJ, Saliba RM, et al.: Long-term follow-up of patients with CLL treated with allogeneic hematopoietic transplantation. Cytotherapy 4 (3): 217-21, 2002.
Doney KC, Chauncey T, Appelbaum FR, et al.: Allogeneic related donor hematopoietic stem cell transplantation for treatment of chronic lymphocytic leukemia. Bone Marrow Transplant 29 (10): 817-23, 2002.
Pavletic SZ, Khouri IF, Haagenson M, et al.: Unrelated donor marrow transplantation for B-cell chronic lymphocytic leukemia after using myeloablative conditioning: results from the Center for International Blood and Marrow Transplant research. J Clin Oncol 23 (24): 5788-94, 2005.
Thompson PA, Tam CS, O'Brien SM, et al.: Fludarabine, cyclophosphamide, and rituximab treatment achieves long-term disease-free survival in IGHV-mutated chronic lymphocytic leukemia. Blood 127 (3): 303-9, 2016.
Fischer K, Bahlo J, Fink AM, et al.: Long-term remissions after FCR chemoimmunotherapy in previously untreated patients with CLL: updated results of the CLL8 trial. Blood 127 (2): 208-15, 2016.
Rossi D, Terzi-di-Bergamo L, De Paoli L, et al.: Molecular prediction of durable remission after first-line fludarabine-cyclophosphamide-rituximab in chronic lymphocytic leukemia. Blood 126 (16): 1921-4, 2015.
Hallek M, Cheson BD, Catovsky D, et al.: iwCLL guidelines for diagnosis, indications for treatment, response assessment, and supportive management of CLL. Blood 131 (25): 2745-2760, 2018.
Burger JA, Tedeschi A, Barr PM, et al.: Ibrutinib as Initial Therapy for Patients with Chronic Lymphocytic Leukemia. N Engl J Med 373 (25): 2425-37, 2015.
Cramer P, Tausch E, von Tresckow J, et al.: Durable remissions following combined targeted therapy in patients with CLL harboring TP53 deletions and/or mutations. Blood 138 (19): 1805-1816, 2021.
Eichhorst B, Fink AM, Bahlo J, et al.: First-line chemoimmunotherapy with bendamustine and rituximab versus fludarabine, cyclophosphamide, and rituximab in patients with advanced chronic lymphocytic leukaemia (CLL10): an international, open-label, randomised, phase 3, non-inferiority trial. Lancet Oncol 17 (7): 928-942, 2016.
Intravenous immunoglobulin for the prevention of infection in chronic lymphocytic leukemia. A randomized, controlled clinical trial. Cooperative Group for the Study of Immunoglobulin in Chronic Lymphocytic Leukemia. N Engl J Med 319 (14): 902-7, 1988.
Griffiths H, Brennan V, Lea J, et al.: Crossover study of immunoglobulin replacement therapy in patients with low-grade B-cell tumors. Blood 73 (2): 366-8, 1989.
Weeks JC, Tierney MR, Weinstein MC: Cost effectiveness of prophylactic intravenous immune globulin in chronic lymphocytic leukemia. N Engl J Med 325 (2): 81-6, 1991.
Mato AR, Roeker LE, Lamanna N, et al.: Outcomes of COVID-19 in patients with CLL: a multicenter international experience. Blood 136 (10): 1134-1143, 2020.
Scarfò L, Chatzikonstantinou T, Rigolin GM, et al.: COVID-19 severity and mortality in patients with chronic lymphocytic leukemia: a joint study by ERIC, the European Research Initiative on CLL, and CLL Campus. Leukemia 34 (9): 2354-2363, 2020.
Mauro FR, Foa R, Cerretti R, et al.: Autoimmune hemolytic anemia in chronic lymphocytic leukemia: clinical, therapeutic, and prognostic features. Blood 95 (9): 2786-92, 2000.
Kaufman M, Limaye SA, Driscoll N, et al.: A combination of rituximab, cyclophosphamide and dexamethasone effectively treats immune cytopenias of chronic lymphocytic leukemia. Leuk Lymphoma 50 (6): 892-9, 2009.
Cheson BD, Frame JN, Vena D, et al.: Tumor lysis syndrome: an uncommon complication of fludarabine therapy of chronic lymphocytic leukemia. J Clin Oncol 16 (7): 2313-20, 1998.
Maddocks-Christianson K, Slager SL, Zent CS, et al.: Risk factors for development of a second lymphoid malignancy in patients with chronic lymphocytic leukaemia. Br J Haematol 139 (3): 398-404, 2007.
Visentin A, Bonaldi L, Rigolin GM, et al.: The complex karyotype landscape in chronic lymphocytic leukemia allows the refinement of the risk of Richter syndrome transformation. Haematologica 107 (4): 868-876, 2022.
Rossi D, Spina V, Deambrogi C, et al.: The genetics of Richter syndrome reveals disease heterogeneity and predicts survival after transformation. Blood 117 (12): 3391-401, 2011.
Chigrinova E, Rinaldi A, Kwee I, et al.: Two main genetic pathways lead to the transformation of chronic lymphocytic leukemia to Richter syndrome. Blood 122 (15): 2673-82, 2013.
Parry EM, Leshchiner I, Guièze R, et al.: Evolutionary history of transformation from chronic lymphocytic leukemia to Richter syndrome. Nat Med 29 (1): 158-169, 2023.
Parry EM, Ten Hacken E, Wu CJ: Richter syndrome: novel insights into the biology of transformation. Blood 142 (1): 11-22, 2023.
Robertson LE, Pugh W, O'Brien S, et al.: Richter's syndrome: a report on 39 patients. J Clin Oncol 11 (10): 1985-9, 1993.
Jain N, Keating M, Thompson P, et al.: Ibrutinib and Venetoclax for First-Line Treatment of CLL. N Engl J Med 380 (22): 2095-2103, 2019.
Ben-Dali Y, Hleuhel MH, da Cunha-Bang C, et al.: Richter's transformation in patients with chronic lymphocytic leukaemia: a Nationwide Epidemiological Study. Leuk Lymphoma 61 (6): 1435-1444, 2020.
Stephens DM, Boucher K, Kander E, et al.: Hodgkin lymphoma arising in patients with chronic lymphocytic leukemia: outcomes from a large multi-center collaboration. Haematologica 106 (11): 2845-2852, 2021.
Al-Sawaf O, Robrecht S, Bahlo J, et al.: Richter transformation in chronic lymphocytic leukemia (CLL)-a pooled analysis of German CLL Study Group (GCLLSG) front line treatment trials. Leukemia 35 (1): 169-176, 2021.
Abdel-Qadir H, Sabrie N, Leong D, et al.: Cardiovascular Risk Associated With Ibrutinib Use in Chronic Lymphocytic Leukemia: A Population-Based Cohort Study. J Clin Oncol 39 (31): 3453-3462, 2021.
Byrd JC, Hillmen P, Ghia P, et al.: Acalabrutinib Versus Ibrutinib in Previously Treated Chronic Lymphocytic Leukemia: Results of the First Randomized Phase III Trial. J Clin Oncol 39 (31): 3441-3452, 2021.
Treatment of Asymptomatic Chronic Lymphocytic Leukemia
Treatment Options for Asymptomatic Chronic Lymphocytic Leukemia (CLL)
Observation
Because of its indolent nature, chemotherapy is not indicated for asymptomatic or minimally affected patients with CLL, and observation is the generally accepted approach.[1] Because the rate of progression may vary, with long periods of stability and sometimes spontaneous regressions, frequent and careful observation is required to monitor the clinical course. One nomogram to predict time-to-first treatment relies on the number of lymph node sites, size of cervical lymph nodes, lactate dehydrogenase level, the IGH mutational status, and the presence of del(11q) or del(17p) established by fluorescence in situ hybridization analysis.[2] Spontaneous regression, manifested by a sustained reduction of the malignant clone without therapy, occurs in less than 5% of patients. These patients almost exclusively have hypermutation of IGH.[3]
Evidence (observation):
The French Cooperative Group on Chronic Lymphocytic Leukemia randomly assigned 1,535 patients with previously untreated stage A disease to receive either chlorambucil or no immediate treatment.[4]
The results showed no survival advantage for immediate treatment with chlorambucil.[4][Level of evidence A1]
A meta-analysis evaluated six trials of patients with early-stage CLL that involved immediate versus deferred therapy with chlorambucil (including the aforementioned trial by the French Cooperative Group).[5]
The results showed no difference in overall survival at 10 years.[5][Level of evidence A1]
Despite many therapeutic options, observation should be considered for asymptomatic or minimally affected patients, even in the context of adverse prognostic findings. Therapy begins when patients develop profound cytopenias or when symptoms adversely impact quality of life.
There are no clinical trial results that confirm that immediate treatment of asymptomatic or minimally affected patients with the B-cell receptor inhibitors or BCL2 inhibitors is superior to observation.
Clinical trials will need to establish improved outcomes using the newer biological therapies in asymptomatic patients before observation or watchful waiting is discontinued.
Current Clinical Trials
Use our advanced clinical trial search to find NCI-supported cancer clinical trials that are now enrolling patients. The search can be narrowed by location of the trial, type of treatment, name of the drug, and other criteria. General information about clinical trials is also available.
References:
Casper JT: Prognostic features of early chronic lymphocytic leukaemia. International Workshop on CLL. Lancet 2 (8669): 968-9, 1989.
Molica S, Giannarelli D, Gentile M, et al.: External validation on a prospective basis of a nomogram for predicting the time to first treatment in patients with chronic lymphocytic leukemia. Cancer 119 (6): 1177-85, 2013.
Kwok M, Oldreive C, Rawstron AC, et al.: Integrative analysis of spontaneous CLL regression highlights genetic and microenvironmental interdependency in CLL. Blood 135 (6): 411-428, 2020.
Dighiero G, Maloum K, Desablens B, et al.: Chlorambucil in indolent chronic lymphocytic leukemia. French Cooperative Group on Chronic Lymphocytic Leukemia. N Engl J Med 338 (21): 1506-14, 1998.
Chemotherapeutic options in chronic lymphocytic leukemia: a meta-analysis of the randomized trials. CLL Trialists' Collaborative Group. J Natl Cancer Inst 91 (10): 861-8, 1999.
Treatment of Symptomatic or Progressive Chronic Lymphocytic Leukemia
Treatment Options for Symptomatic or Progressive Chronic Lymphocytic Leukemia (CLL)
The following regimens are considered first-line treatment approaches for patients with CLL who are experiencing symptomatic progression:
R-CHOP (rituximab, cyclophosphamide, doxorubicin, vincristine, and prednisone) (only if Richter syndrome with histological progression is suspected clinically).
Several large prospective clinical trials have compared these approaches. A chemotherapy-free approach for first-line therapy is usually preferred for most patients, but it is mandatory for patients with del(17p) or TP53-altered disease.[1,2,3,4,5]
BTK inhibitors
Ibrutinib versus zanubrutinib
Evidence (ibrutinib vs. zanubrutinib):
A prospective randomized trial of 652 patients with relapsed or refractory CLL compared ibrutinib and zanubrutinib.[6]
With a median follow-up of 29.6 months, the 2-year progression-free survival (PFS) rate was 78.4% in the zanubrutinib group and 65.9% in the ibrutinib group (hazard ratio [HR], 0.65; 95% confidence interval [CI], 0.49–0.86; P = .002).[6][Level of evidence B1]
Cardiac disorders leading to treatment discontinuation occurred in 1 patient (0.3%) in the zanubrutinib group and 14 patients (4.3%) in the ibrutinib group. Six deaths caused by cardiac events were all in patients who received ibrutinib.
Ibrutinib versus acalabrutinib
Evidence (ibrutinib vs. acalabrutinib):
A prospective randomized trial of 533 previously untreated patients compared the BTK inhibitors acalabrutinib and ibrutinib.[7]
With a median follow-up of 41 months, acalabrutinib was noninferior to ibrutinib, with a median PFS of 38.4 months for patients in both arms of the trial (HR, 1.00; 95% CI, 0.79–1.27).[7][Level of evidence B1]
The incidence of atrial fibrillation of any grade was lower for patients who received acalabrutinib than for patients who received ibrutinib (9.4% vs. 16.0%, P = .02). In this trial, the incidence of diarrhea and headaches was significantly higher in patients who received acalabrutinib (P < .05), while musculoskeletal pain was higher in patients who received ibrutinib (P = .0229).[8]
Ventricular arrhythmias and sudden death events appear to be a side effect of all the BTK inhibitors. For acalabrutinib, these events occur with a relative risk of 8.2 over age-matched controls.[9]
Ibrutinib versus ibrutinib plus rituximab versus BR
Evidence (ibrutinib vs. ibrutinib plus rituximab vs. BR):
A prospective trial included 547 previously untreated patients aged 65 years or older. Patients were randomly assigned to receive BR, ibrutinib alone, or ibrutinib plus rituximab.[10]
With a median follow-up of 38 months, the 2-year PFS rate was 74% for patients who received BR. The rates were significantly higher for patients who received ibrutinib alone (87%; HR, 0.39; 95% CI, 0.25–0.58) or ibrutinib plus rituximab (88%; HR, 0.38; 95% CI, 0.25–0.59; P < .001).[10][Level of evidence B1]
There was no difference in PFS between the two ibrutinib groups (HR, 1.00; 95% CI, 0.62–1.62; P = .49) and no difference in overall survival (OS) between each of the groups.
Ibrutinib versus rituximab plus ibrutinib
Evidence (ibrutinib vs. rituximab plus ibrutinib):
A prospective randomized trial included 208 patients who were previously untreated or had relapsed disease. Patients received rituximab plus ibrutinib or ibrutinib alone.[11]
With a median follow-up of 36 months, there was no difference in PFS (86%; HR, 1.04; 95% CI, 0.49−2.20; P = .91).[11][Level of evidence B1]
Ibrutinib versus FCR
Ibrutinib is a selective irreversible inhibitor of BTK, a signaling molecule located upstream in the B-cell receptor-signaling cascade.
Evidence (ibrutinib vs. FCR):
ECOG-E1912 (NCT02048813) was a prospective randomized trial that compared ibrutinib and rituximab with FCR. A total of 529 patients previously untreated for CLL were randomly assigned in a 2:1 ratio to receive either ibrutinib and rituximab followed by ibrutinib maintenance (354 patients) or six cycles of FCR (175 patients).[12,13]
With a median follow-up of 5.8 years, the 5-year OS rate favored the ibrutinib arm (95% vs. 89%) (HR, 0.47; 95% CI, 0.25–0.89; P = .018).[13][Level of evidence A1]
In 281 patients without IGH pathogenic variants, the 5-year PFS rate favored ibrutinib plus rituximab versus FCR (75% vs. 33%) (HR, 0.27; 95% CI, 0.18−0.41; P < .0001). In 114 patients with IGH pathogenic variants, the 5-year PFS rate was also significantly different, at 83% for the ibrutinib arm and 68% for the FCR arm (HR, 0.27; 95% CI, 0.11−0.62; P = .001).[12,13,14]
Although undetectable measurable residual disease (MRD) was less than 10% between 12 and 36 months follow-up, patients with detectable MRD did not have significantly worse PFS (P = .14 at 12 months, P = .90 at 24 months, and P = .53 at 36 months).[15]
Zanubrutinib versus BR
Evidence (zanubrutinib vs. BR):
A prospective randomized trial included 590 previously untreated patients aged 65 years or older. Patients were randomly assigned to receive zanubrutinib or BR.[16]
With a median follow-up of 26.2 months, the 2-year PFS rate was 85.5% (95% CI, 80.1%–89.6%) for patients who received zanubrutinib and 69.5% (95% CI, 62.4%–75.5%) for patients who received BR (HR, 0.42; 95% CI, 0.28–0.63; P < .0001).[16][Level of evidence B1]
Acalabrutinib plus obinutuzumab versus acalabrutinib versus chlorambucil plus obinutuzumab
Acalabrutinib is a highly selective covalent irreversible BTK inhibitor, designed to minimize the gastrointestinal toxicities and risk of atrial fibrillation seen with ibrutinib.
Evidence (acalabrutinib plus obinutuzumab vs. acalabrutinib vs. chlorambucil plus obinutuzumab):
The prospective ELEVATE TN trial (NCT02475681) included 535 previously untreated patients aged 65 years or older with comorbidities (e.g., creatinine clearance <70 mL/min). Patients were randomly assigned to one of three arms: acalabrutinib plus obinutuzumab, acalabrutinib alone, or chlorambucil plus obinutuzumab.[17]
With a median follow-up of 28 months, the 24-month PFS rates were 93% for acalabrutinib plus obinutuzumab (HR, 0.10; 95% CI, 0.06−0.17; P < .0001), 87% for acalabrutinib alone (HR, 0.20; 95% CI, 0.13−0.30; P < .0001), and 47% for chlorambucil plus obinutuzumab.[17][Level of evidence B1]
There was a small but significant difference in the PFS rates between the two acalabrutinib groups (93% vs. 87% at 24 months), which favored the combination (HR, 0.49; 95% CI, 0.26−0.95).[17]
Venetoclax with initial use of obinutuzumab and rituximab
Venetoclax plus obinutuzumab versus chlorambucil plus obinutuzumab
Evidence (venetoclax plus obinutuzumab vs. chlorambucil plus obinutuzumab):
The prospective CLL14 trial (NCT02242942) included 432 previously untreated patients with significant medical comorbidities (6 or higher on the Cumulative Illness Rating Scale; median age, 72 years). Patients were randomly assigned to receive either venetoclax (a highly selective inhibitor of BCL2) plus obinutuzumab (the human anti-CD20 monoclonal antibody) or chlorambucil plus obinutuzumab.[18]
With a median follow-up of 76.4 months, the median PFS rate was 76.2 months for venetoclax plus obinutuzumab, compared with 36.4 months for chlorambucil plus obinutuzumab (HR, 0.40; 95% CI, 0.31–0.52; P < .0001).[18][Level of evidence B1]
The 6-year OS rate was 78.7% for venetoclax plus obinutuzumab and 69.2% for chlorambucil plus obinutuzumab (HR, 0.69; 95% CI, 0.48–1.01; P = .052).
Venetoclax plus rituximab (VenR) versus BR
Evidence (VenR vs. BR):
The prospective MURANO trial (NCT02005471) included 389 patients with relapsed or refractory CLL. Patients were randomly assigned to receive either VenR (venetoclax for 2 years plus rituximab for the first 6 months) or 6 months of BR.
With a median follow-up of 24 months, the 2-year PFS rate was 84.9% for VenR and 36.3% for BR (HR, 0.17; 95% CI, 0.11–0.25; P < .001).[19,20] An update with a 48-month median follow-up, reported in abstract form, showed a 4-year OS rate of 85.3% for VenR and 66.8% for BR (HR, 0.41; 95% CI, 0.26−0.65; P < .0001).[21][Level of evidence A1]
With a median follow-up of 59.2 months, the 5-year OS rate was 82.1% (95% CI, 76.4%–87.8%) for patients who received VenR and 62.2% (95% CI, 54.8%–69.6%) for patients who received BR (P < .0001).[22][Level of evidence A1] The median time to next treatment was 57.8 months (95% CI, 55.1–not estimable) for VenR.
For the 43% of patients assigned to VenR who achieved undetectable MRD at the end of treatment, the median time to MRD conversion was 19.4 months. The median time from MRD conversion to overt clinical progressive disease was another 25.2 months.[22]
Bendamustine and rituximab
BR versus FCR
Evidence (BR vs. FCR):
The German CLL Study Group compared BR versus FCR as first-line therapy in patients with CLL who required therapy.[23]
With a median follow-up of 37.1 months, the median PFS was better for patients who received FCR (55.2 months vs. 41.7 months) (HR, 1.64; 90% CI, 1.31–2.06; P = .001), but there was no difference in the OS rate at 3 years (91% vs. 92%, not significant).[23][Level of evidence B1]
In patients older than 65 years, there was no difference in PFS between the two arms, but more infections occurred with FCR than with BR (grade 3 to 5 infection, 47% vs. 27%).
Fludarabine, cyclophosphamide, and rituximab (FCR)
FCR
FCR is used for patients with an IGH hypermutation.
Evidence (FCR):
Several trials used FCR for appropriate patients with IGH pathogenic variants who required therapy.
The PFS rate exceeded 60% at more than 10 years.[24,25,26][Level of evidence C2] Nonetheless, late relapses were seen beyond 10 years.
BTK inhibitor plus venetoclax
BTK inhibitor (ibrutinib or acalabrutinib) plus venetoclax
Evidence (BTK inhibitor [ibrutinib or acalabrutinib] plus venetoclax):
A prospective randomized trial included 523 patients with previously untreated CLL. Patients received either ibrutinib plus venetoclax for up to 6 years or FCR for six cycles.[27] This comparison was embedded in a larger three-arm randomization that also included ibrutinib alone for up to 6 years; the statistics were thought too preliminary to analyze the ibrutinib-alone group versus the other arms.
With a median follow-up of 43.7 months, the 3-year OS rate was 98.0% (95% CI, 95.2%–99.2%) in the ibrutinib-plus-venetoclax group and 93.0% (95% CI, 88.9%–95.6%) in the FCR group (HR, 0.31; 95% CI, 0.15–0.67).[27][Level of evidence A1]
The 3-year OS benefit favored ibrutinib plus venetoclax (HR, 0.23; 95% CI, 0.06–0.81) for patients without IGH pathogenic variants, but not for those with IGH pathogenic variants (HR, 0.61; 95% CI, 0.20–1.82).
The 3-year PFS rate was 97.2% (95% CI, 94.1%–98.6%) in the ibrutinib-plus-venetoclax group and 76.8% (95% CI, 70.8%–81.7%) in the FCR group (HR, 0.13; 95% CI, 0.07–0.24; P < .001).
MRD negativity was a requirement for stopping therapy with venetoclax and ibrutinib after 2 years. This trial did not randomly assign patients to either continued therapy or cessation of therapy based on MRD status. The utility and value of MRD could not be determined from this trial due to its design.
A prospective randomized trial (GLOW [NCT03462719]) included 211 patients with previously untreated CLL. Patients were aged 65 and older or aged 18 to 64 with comorbidities and an Eastern Cooperative Oncology Group performance status of 0 to 2. Patients were randomly assigned to receive either fixed-duration ibrutinib and venetoclax (15 months total) or chlorambucil and obinutuzumab (6 months total).[28]
With a median follow-up of 46 months, the 42-month PFS rate was 74.6% in the ibrutinib-plus-venetoclax group and 24.8% in the chlorambucil-plus-obinutuzumab group (HR, 0.214; 95% CI, 0.138–0.334; P < .0001).[28][Level of evidence B1]
Ibrutinib was given for 3 months prior to the combination of venetoclax with ibrutinib to avoid tumor lysis syndrome.
At 3 months after the end of therapy, 40.6% of patients who received ibrutinib and venetoclax achieved undetectable MRD (<10-5 by next-generation sequencing) in the bone marrow, and 43.4% of patients achieved undetectable MRD in the peripheral blood.[28] However, the PFS rate exceeded 90% at 12 months after the end of therapy regardless of if the MRD was detectable or not.
A prospective trial included 867 patients with previously untreated CLL. Patients were randomly assigned to one of three treatment regimens: (1) AV (acalabrutinib and venetoclax), (2) AVO (acalabrutinib, venetoclax, and obinutuzumab), or (3) either BR or FCR (at the discretion of the investigator).[29]
With a median follow-up of 41 months, the 3-year OS rate was 94.1% for patients who received AV and 85.9% for patients who received BR or FCR (HR, 0.33; 95% CI, 0.18–0.56; P < .0001). However, the 3-year OS rate was 87.7% for patients who received AVO compared with 85.9% for patients who received BR or FCR (HR, 0.76; 95% CI, 0.49–1.18; P = nonsignificant).[29][Level of evidence A1]
The 3-year PFS rate was 76.5% for patients who received AV, 83.1% for patients who received AVO, and 66.5% for patients who received BR or FCR. The PFS for patients who received AV versus those who received BR or FCR had an HR of 0.65 (95% CI, 0.49–0.87; P = .004). The PFS for patients who received AVO versus those who received BR or FCR had an HR of 0.42 (95% CI, 0.30–0.59; P < .001).
Although treatment with AVO resulted in a better PFS than AV, there were increased infectious deaths with AVO, especially from COVID-19 (SARS-CoV-2). These deaths accounted for a nonsignificant difference in OS rates between patients who received AVO (87.7%) and patients who received BR or FCR (85.9%).
A phase II trial included 80 previously untreated patients who were aged 65 or older or had high-risk disease. Patients were treated with ibrutinib for 3 months followed by combined ibrutinib plus venetoclax for a total of 24 months.[30]
With a median follow-up of 38.5 months, the 2-year response rate was 82%, the 3-year PFS rate was 93% (95% CI, 88%–99%), and the 3-year OS rate was 96%.[30][Level of evidence C1]
The toxicity of the combination is similar to either agent alone and avoids the tumor lysis syndrome seen when starting with venetoclax.
High rates of undetectable MRD (<10-4 by 8-color flow cytometry) in the peripheral blood (75%) and bone marrow (72%) is unprecedented. The clinical significance of this finding awaits prospective randomized trials to establish clinical outcomes versus either drug alone. This combination has also been tested in the relapsed/refractory setting.[31]
A phase II trial included 159 previously untreated patients aged 70 years or older. Patients received 3 cycles of ibrutinib alone followed by 12 cycles of ibrutinib plus venetoclax.[32]
With a median follow-up of 27.9 months, the complete response rate was 55% (95% CI, 48%–63%), the undetectable MRD rate was 77% (95% CI, 70%–83%) for blood and 60% (95% CI, 52%–67%) for bone marrow, the 2-year PFS rate was 95% (95% CI, 90%–97%), and the 2-year OS rate was 98% (95% CI, 94%–99%).[32][Level of evidence C1]
The toxicity of the ibrutinib-venetoclax combination was similar to either agent alone, and the ibrutinib-alone lead-in avoided tumor lysis syndrome.
A phase II trial included 41 previously untreated patients who received ibrutinib plus venetoclax and obinutuzumab. Patients with undetectable MRD at cycle 16 could opt to discontinue treatment if they had a complete response.[33]
With a median follow-up of 38.4 months, the 3-year PFS rate was 79.9% and the 3-year OS rate was 92.6%.[33][Level of evidence C1]
In summary, these trials establish the use of venetoclax with obinutuzumab or rituximab, or the use of ibrutinib, acalabrutinib, or zanubrutinib as first-line therapy in patients with previously untreated CLL. A lower rate of atrial fibrillation occurs with acalabrutinib or zanubrutinib than with ibrutinib.[6,7,34,35] Unlike ibrutinib or acalabrutinib, which are given continuously until relapse, venetoclax may be stopped after 12 months, with durable maintenance of remission. Venetoclax, ibrutinib, acalabrutinib, or zanubrutinib can be readministered with success, if needed.[35,36] These targeting drugs are also effective for patients with TP53 pathogenic variants.[37] Different regimens of these drugs, as standalone agents or in combinations, with or without obinutuzumab, need to be evaluated in prospective randomized trials. Several provocative phase II and III trials with these combinations have resulted in unprecedented rates of MRD-negative disease which appear more durable;[30,38,39,40,41] whether this results in any clinical advantage to a more sequential approach requires prospective randomized trials. The National Comprehensive Cancer Network has listed the combination of venetoclax and ibrutinib as a first-line treatment option for patients with previously untreated CLL.[42] The considerable financial toxicity of these combinations mandates verification of superior efficacy. These trials further establish the rationale for a chemotherapy-free approach for first-line therapy for CLL instead of the previous standard of BR and FCR (which proved more efficacious than chlorambucil regimens). Patients at the highest risk of relapse have multiple poor prognostic factors, including TP53 pathogenic variants, elevated lactate dehydrogenase, elevated beta-2 microglobulin, and prior treatment.[43] These highest risk patients should consider clinical trials.
MRD Testing Outside the Context of a Clinical Trial
Undetectable MRD (≤1 × 10-4 CLL cells in peripheral blood or bone marrow aspirates) can be confirmed by flow cytometry or next-generation sequencing. The attainment of undetectable MRD represents an even more stringent complete remission that was prognostic for improved PFS and OS in multiple studies.[22,44,45,46]The use of time-limited therapy with venetoclax, or the investigational combination of venetoclax and a BTK inhibitor, has produced complete responses, and even undetectable MRD, in most patients.[22,44,47]
Testing for undetectable MRD has become a standard parameter for defining responses in all modern clinical trials for CLL. Undetectable MRD has prognostic value but its status as a predictive marker is uncertain. The potential value of MRD testing in routine clinical practice depends on whether it can be used for clinical decision making such as stopping, changing, or continuing treatment. High-level evidence for this intervention would require a prospective randomized clinical trial in which MRD was used as a predictive biomarker for a group attaining an OS advantage compared with a control group disregarding MRD status. Such evidence has not been attained. Similar to outcomes in follicular lymphoma and other indolent lymphoid neoplasms, improved PFS in patients with CLL does not directly predict OS.
A phase II trial used a combination of venetoclax and ibrutinib in patients with undetectable MRD after 1 year of therapy. Patients were randomly assigned to receive either ibrutinib maintenance therapy or treatment cessation.[47] All MRD-positive patients continued to receive ibrutinib. With a median follow-up of 34.4 months, the 1-year PFS rates were 98% for patients with undetectable MRD who ceased therapy, 96% for patients with undetectable MRD who received continued ibrutinib, and 97% for MRD-positive patients who received continued ibrutinib. All patients did well at 12 months. Knowledge of MRD status to guide discontinuation of therapy did not affect the outcome.[47]
Another phase II trial included 70 previously untreated patients who received 1 year of venetoclax plus obinutuzumab. Patients were randomly assigned to receive another year of venetoclax irrespective of MRD status, or another year of venetoclax only if they were MRD-positive. With a median follow-up of 35.2 months, the MRD rates were identical for both randomized groups, suggesting no improved efficacy with this approach, although increased adverse events were reported with an extra year of venetoclax.[48]
Before MRD is used outside the context of a clinical trial, conclusive evidence is required to establish that MRD is a predictive biomarker that can guide clinical decisions.
Current Clinical Trials
Use our advanced clinical trial search to find NCI-supported cancer clinical trials that are now enrolling patients. The search can be narrowed by location of the trial, type of treatment, name of the drug, and other criteria. General information about clinical trials is also available.
References:
Byrd JC, Furman RR, Coutre SE, et al.: Targeting BTK with ibrutinib in relapsed chronic lymphocytic leukemia. N Engl J Med 369 (1): 32-42, 2013.
O'Brien S, Furman RR, Coutre S, et al.: Single-agent ibrutinib in treatment-naïve and relapsed/refractory chronic lymphocytic leukemia: a 5-year experience. Blood 131 (17): 1910-1919, 2018.
O'Brien S, Jones JA, Coutre SE, et al.: Ibrutinib for patients with relapsed or refractory chronic lymphocytic leukaemia with 17p deletion (RESONATE-17): a phase 2, open-label, multicentre study. Lancet Oncol 17 (10): 1409-1418, 2016.
Stilgenbauer S, Eichhorst B, Schetelig J, et al.: Venetoclax in relapsed or refractory chronic lymphocytic leukaemia with 17p deletion: a multicentre, open-label, phase 2 study. Lancet Oncol 17 (6): 768-78, 2016.
Stilgenbauer S, Eichhorst B, Schetelig J, et al.: Venetoclax for Patients With Chronic Lymphocytic Leukemia With 17p Deletion: Results From the Full Population of a Phase II Pivotal Trial. J Clin Oncol 36 (19): 1973-1980, 2018.
Brown JR, Eichhorst B, Hillmen P, et al.: Zanubrutinib or Ibrutinib in Relapsed or Refractory Chronic Lymphocytic Leukemia. N Engl J Med 388 (4): 319-332, 2023.
Byrd JC, Hillmen P, Ghia P, et al.: Acalabrutinib Versus Ibrutinib in Previously Treated Chronic Lymphocytic Leukemia: Results of the First Randomized Phase III Trial. J Clin Oncol 39 (31): 3441-3452, 2021.
Seymour JF, Byrd JC, Ghia P, et al.: Detailed safety profile of acalabrutinib vs ibrutinib in previously treated chronic lymphocytic leukemia in the ELEVATE-RR trial. Blood 142 (8): 687-699, 2023.
Bhat SA, Gambril J, Azali L, et al.: Ventricular arrhythmias and sudden death events following acalabrutinib initiation. Blood 140 (20): 2142-2145, 2022.
Woyach JA, Ruppert AS, Heerema NA, et al.: Ibrutinib Regimens versus Chemoimmunotherapy in Older Patients with Untreated CLL. N Engl J Med 379 (26): 2517-2528, 2018.
Burger JA, Sivina M, Jain N, et al.: Randomized trial of ibrutinib vs ibrutinib plus rituximab in patients with chronic lymphocytic leukemia. Blood 133 (10): 1011-1019, 2019.
Shanafelt TD, Wang XV, Kay NE, et al.: Ibrutinib-Rituximab or Chemoimmunotherapy for Chronic Lymphocytic Leukemia. N Engl J Med 381 (5): 432-443, 2019.
Shanafelt TD, Wang XV, Hanson CA, et al.: Long-term outcomes for ibrutinib-rituximab and chemoimmunotherapy in CLL: updated results of the E1912 trial. Blood 140 (2): 112-120, 2022.
Shanafelt TD, Wang V, Kay NE, et al.: Ibrutinib and rituximab provides superior clinical outcome compared to FCR in younger patients with chronic lymphocytic leukemia (CLL): extended follow-up from the E1912 trial. [Abstract] Blood 134 (Suppl 1): A-33, 2019.
Wang XV, Hanson CA, Tschumper RC, et al.: Measurable residual disease does not preclude prolonged progression-free survival in CLL treated with ibrutinib. Blood 138 (26): 2810-2827, 2021.
Tam CS, Brown JR, Kahl BS, et al.: Zanubrutinib versus bendamustine and rituximab in untreated chronic lymphocytic leukaemia and small lymphocytic lymphoma (SEQUOIA): a randomised, controlled, phase 3 trial. Lancet Oncol 23 (8): 1031-1043, 2022.
Sharman JP, Egyed M, Jurczak W, et al.: Acalabrutinib with or without obinutuzumab versus chlorambucil and obinutuzmab for treatment-naive chronic lymphocytic leukaemia (ELEVATE TN): a randomised, controlled, phase 3 trial. Lancet 395 (10232): 1278-1291, 2020.
Al-Sawaf O, Robrecht S, Zhang C, et al.: Venetoclax-obinutuzumab for previously untreated chronic lymphocytic leukemia: 6-year results of the randomized phase 3 CLL14 study. Blood 144 (18): 1924-1935, 2024.
Seymour JF, Kipps TJ, Eichhorst B, et al.: Venetoclax-Rituximab in Relapsed or Refractory Chronic Lymphocytic Leukemia. N Engl J Med 378 (12): 1107-1120, 2018.
Kater AP, Seymour JF, Hillmen P, et al.: Fixed Duration of Venetoclax-Rituximab in Relapsed/Refractory Chronic Lymphocytic Leukemia Eradicates Minimal Residual Disease and Prolongs Survival: Post-Treatment Follow-Up of the MURANO Phase III Study. J Clin Oncol 37 (4): 269-277, 2019.
Seymour JF, Kipps TJ, Eichhorst BF, et al.: Four-year analysis of Murano study confirms sustained benefit of time-limited venetoclax-rituximab (VenR) in relapsed/refractory (R/R) chronic lymphocytic leukemia (CLL). [Abstract] Blood 134 (Suppl 1): A-355, 2019.
Seymour JF, Kipps TJ, Eichhorst BF, et al.: Enduring undetectable MRD and updated outcomes in relapsed/refractory CLL after fixed-duration venetoclax-rituximab. Blood 140 (8): 839-850, 2022.
Eichhorst B, Fink AM, Bahlo J, et al.: First-line chemoimmunotherapy with bendamustine and rituximab versus fludarabine, cyclophosphamide, and rituximab in patients with advanced chronic lymphocytic leukaemia (CLL10): an international, open-label, randomised, phase 3, non-inferiority trial. Lancet Oncol 17 (7): 928-942, 2016.
Thompson PA, Tam CS, O'Brien SM, et al.: Fludarabine, cyclophosphamide, and rituximab treatment achieves long-term disease-free survival in IGHV-mutated chronic lymphocytic leukemia. Blood 127 (3): 303-9, 2016.
Fischer K, Bahlo J, Fink AM, et al.: Long-term remissions after FCR chemoimmunotherapy in previously untreated patients with CLL: updated results of the CLL8 trial. Blood 127 (2): 208-15, 2016.
Rossi D, Terzi-di-Bergamo L, De Paoli L, et al.: Molecular prediction of durable remission after first-line fludarabine-cyclophosphamide-rituximab in chronic lymphocytic leukemia. Blood 126 (16): 1921-4, 2015.
Munir T, Cairns DA, Bloor A, et al.: Chronic Lymphocytic Leukemia Therapy Guided by Measurable Residual Disease. N Engl J Med 390 (4): 326-337, 2024.
Niemann CU, Munir T, Moreno C, et al.: Fixed-duration ibrutinib-venetoclax versus chlorambucil-obinutuzumab in previously untreated chronic lymphocytic leukaemia (GLOW): 4-year follow-up from a multicentre, open-label, randomised, phase 3 trial. Lancet Oncol 24 (12): 1423-1433, 2023.
Brown JR, Seymour JF, Jurczak W, et al.: Fixed-Duration Acalabrutinib Combinations in Untreated Chronic Lymphocytic Leukemia. N Engl J Med 392 (8): 748-762, 2025.
Wierda WG, Allan JN, Siddiqi T, et al.: Ibrutinib Plus Venetoclax for First-Line Treatment of Chronic Lymphocytic Leukemia: Primary Analysis Results From the Minimal Residual Disease Cohort of the Randomized Phase II CAPTIVATE Study. J Clin Oncol 39 (34): 3853-3865, 2021.
Hillmen P, Rawstron AC, Brock K, et al.: Ibrutinib Plus Venetoclax in Relapsed/Refractory Chronic Lymphocytic Leukemia: The CLARITY Study. J Clin Oncol 37 (30): 2722-2729, 2019.
Tam CS, Allan JN, Siddiqi T, et al.: Fixed-duration ibrutinib plus venetoclax for first-line treatment of CLL: primary analysis of the CAPTIVATE FD cohort. Blood 139 (22): 3278-3289, 2022.
Huber H, Tausch E, Schneider C, et al.: Final analysis of the CLL2-GIVe trial: obinutuzumab, ibrutinib, and venetoclax for untreated CLL with del(17p)/TP53mut. Blood 142 (11): 961-972, 2023.
Hillmen P, Eichhorst B, Brown JR, et al.: Zanubrutinib Versus Ibrutinib in Relapsed/Refractory Chronic Lymphocytic Leukemia and Small Lymphocytic Lymphoma: Interim Analysis of a Randomized Phase III Trial. J Clin Oncol 41 (5): 1035-1045, 2023.
Shadman M, Flinn IW, Levy MY, et al.: Zanubrutinib in patients with previously treated B-cell malignancies intolerant of previous Bruton tyrosine kinase inhibitors in the USA: a phase 2, open-label, single-arm study. Lancet Haematol 10 (1): e35-e45, 2023.
Ma S, Seymour JF, Brander DM, et al.: Efficacy of venetoclax plus rituximab for relapsed CLL: 5-year follow-up of continuous or limited- duration therapy. Blood 138 (10): 836-846, 2021.
Allan JN, Shanafelt T, Wiestner A, et al.: Long-term efficacy of first-line ibrutinib treatment for chronic lymphocytic leukaemia in patients with TP53 aberrations: a pooled analysis from four clinical trials. Br J Haematol 196 (4): 947-953, 2022.
Jain N, Keating M, Thompson P, et al.: Ibrutinib and Venetoclax for First-Line Treatment of CLL. N Engl J Med 380 (22): 2095-2103, 2019.
Rogers KA, Huang Y, Ruppert AS, et al.: Phase II Study of Combination Obinutuzumab, Ibrutinib, and Venetoclax in Treatment-Naïve and Relapsed or Refractory Chronic Lymphocytic Leukemia. J Clin Oncol 38 (31): 3626-3637, 2020.
Soumerai JD, Mato AR, Dogan A, et al.: Zanubrutinib, obinutuzumab, and venetoclax with minimal residual disease-driven discontinuation in previously untreated patients with chronic lymphocytic leukaemia or small lymphocytic lymphoma: a multicentre, single-arm, phase 2 trial. Lancet Haematol 8 (12): e879-e890, 2021.
Huber H, Edenhofer S, von Tresckow J, et al.: Obinutuzumab (GA-101), ibrutinib, and venetoclax (GIVe) frontline treatment for high-risk chronic lymphocytic leukemia. Blood 139 (9): 1318-1329, 2022.
Wierda WG, Brown J, Abramson JS, et al.: Chronic Lymphocytic Leukemia/Small Lymphocytic Lymphoma, Version 2.2024, NCCN Clinical Practice Guidelines in Oncology. J Natl Compr Canc Netw 22 (3): 175-204, 2024.
Ahn IE, Tian X, Ipe D, et al.: Prediction of Outcome in Patients With Chronic Lymphocytic Leukemia Treated With Ibrutinib: Development and Validation of a Four-Factor Prognostic Model. J Clin Oncol 39 (6): 576-585, 2021.
Kovacs G, Robrecht S, Fink AM, et al.: Minimal Residual Disease Assessment Improves Prediction of Outcome in Patients With Chronic Lymphocytic Leukemia (CLL) Who Achieve Partial Response: Comprehensive Analysis of Two Phase III Studies of the German CLL Study Group. J Clin Oncol 34 (31): 3758-3765, 2016.
Al-Sawaf O, Zhang C, Lu T, et al.: Minimal Residual Disease Dynamics after Venetoclax-Obinutuzumab Treatment: Extended Off-Treatment Follow-up From the Randomized CLL14 Study. J Clin Oncol 39 (36): 4049-4060, 2021.
Thompson PA, Srivastava J, Peterson C, et al.: Minimal residual disease undetectable by next-generation sequencing predicts improved outcome in CLL after chemoimmunotherapy. Blood 134 (22): 1951-1959, 2019.
Kater AP, Levin MD, Dubois J, et al.: Minimal residual disease-guided stop and start of venetoclax plus ibrutinib for patients with relapsed or refractory chronic lymphocytic leukaemia (HOVON141/VISION): primary analysis of an open-label, randomised, phase 2 trial. Lancet Oncol 23 (6): 818-828, 2022.
Kersting S, Dubois J, Nasserinejad K, et al.: Venetoclax consolidation after fixed-duration venetoclax plus obinutuzumab for previously untreated chronic lymphocytic leukaemia (HOVON 139/GiVe): primary endpoint analysis of a multicentre, open-label, randomised, parallel-group, phase 2 trial. Lancet Haematol 9 (3): e190-e199, 2022.
Treatment of Recurrent or Refractory Chronic Lymphocytic Leukemia
Treatment Options for Recurrent or Refractory Chronic Lymphocytic Leukemia (CLL)
The same regimens considered for first-line therapy for patients with CLL can be readministered in a sequential fashion. These regimens are described in more detail under first-line therapy. For more information, see the Treatment of Symptomatic or Progressive CLL section.
Bruton tyrosine kinase (BTK) inhibitors (acalabrutinib, zanubrutinib, or ibrutinib).
Venetoclax with initial use of obinutuzumab or rituximab.
Bendamustine and rituximab (BR).
Fludarabine, cyclophosphamide, and rituximab (FCR).
BTK inhibitor (ibrutinib or acalabrutinib) plus venetoclax.
R-CHOP (rituximab, cyclophosphamide, doxorubicin, vincristine, and prednisone) (only if Richter syndrome with histological progression is suspected clinically).
In the relapsed setting, venetoclax showed similar efficacy and safety even after previous therapy with ibrutinib or idelalisib (the phosphatidylinositol 3-kinase [PI3K] delta inhibitor).[1,2]
Similarly, in a trial reported in abstract form, ibrutinib and acalabrutinib showed similar efficacy and safety after previous therapy with venetoclax.[3] Sequencing these novel agents showed efficacy in the relapsed/refractory setting.[4,5]
Noncovalent BTK inhibitors
Unlike other BTK inhibitors (ibrutinib, acalabrutinib, and zanubrutinib), pirtobrutinib binds to BTK in a noncovalent manner.[6]
In a prospective phase I/II study, 317 patients with CLL/small lymphocytic leukemia received pirtobrutinib. A total of 247 of these patients had been treated previously with a covalent-binding BTK inhibitor.[7]
With a median follow-up of 19.4 months, the median progression-free survival (PFS) was 19.6 months (95% confidence interval [CI], 16.9–22.1) for all patients and 16.8 months (95% CI, 13.2–18.7) for the subgroup of patients with BTK inhibitor resistance, intolerance, or BTK C481 variants (a resistance variant for the covalent agents).[7][Level of evidence C3]
In this trial, 82 patients had a diagnosis of Richter transformation. Among those patients, the overall response rate was 50.0% (95% CI, 38.7%–61.3%), and the complete response rate was 13%.[8][Level of evidence C3]
Chimeric antigen receptor (CAR) T-cell therapy
Autologous T cells can be modified by viral vectors to incorporate antigen receptor specificity for the B-cell antigen CD19 and then infused into previously treated patients.[9] A dramatic response lasting 6 months has prompted larger trials of this concept. Ongoing clinical trials are testing the concept of T cells directed at CD19 with engineered CAR T cells.[10,11,12]
PI3K inhibitors
Idelalisib is an oral inhibitor of the delta isoform of PI3K, which is in the B-cell receptor-signaling cascade. This drug has been withdrawn from its U.S. Food and Drug Administration (FDA) indication due to toxicity and is no longer available. Duvelisib is an oral dual inhibitor of the delta and gamma isoforms of PI3K.[13]
In a prospective trial, 319 patients with relapsed and refractory CLL/small lymphocytic lymphoma were randomly assigned to receive duvelisib versus ofatumumab.[14]
With a median follow-up of 22.4 months, the median PFS was significantly higher for patients who received duvelisib, at 13.3 months, than for patients who received ofatumumab, at 9.9 months (hazard ratio [HR], 0.52; P < .0001).[14][Level of evidence B1]
Serious side effects include infections, pneumonitis, diarrhea or colitis, dermatitis, neutropenia, rash, fatigue, pyrexia, nausea, anemia, and transaminitis.
The FDA has approved duvelisib for third-line (or greater) use. However, follow-up data submitted to the FDA showed, with a median follow-up of 63 months, a median OS of 52.3 months (95% CI, 41.8–68.0) in the duvelisib group and 63.3 months (95% CI, 41.2–not estimable) in the ofatumumab group.[15]
Lenalidomide (with or without rituximab)
Lenalidomide is an oral immunomodulatory agent with response rates of more than 50%, with or without rituximab, for patients with previously treated and untreated disease.[16,17,18,19,20,21,22][Level of evidence C3] Prolonged, lower-dose approaches and attention to prevention of tumor lysis syndrome are suggested with this agent.[16,23] Combination therapy and long-term toxicities from using lenalidomide (such as increased myelodysplasia, as seen in myeloma patients) remain undefined for patients with CLL.
Bone marrow or peripheral blood stem cell transplant
In a prospective randomized trial, 241 previously untreated patients younger than 66 years with advanced-stage disease received induction therapy with a CHOP (cyclophosphamide, doxorubicin, vincristine, and prednisone)-based regimen followed by fludarabine.[24] Complete responders (105 patients) were randomly assigned to undergo autologous stem cell transplant (SCT) or observation, while the other 136 patients were randomly assigned to receive dexamethasone, high-dose cytarabine, and cisplatin reinduction followed by either autologous SCT or fludarabine plus cyclophosphamide (FC). Although the 3-year event-free survival (EFS) favored autologous SCT in complete responders, there was no difference in OS in any of the randomized comparisons.[24][Level of evidence B1] Autologous bone marrow/stem cell transplant is rarely employed for patients with relapsed CLL.
Patients with adverse prognostic factors are very likely to die from CLL. These patients are candidates for clinical trials that employ high-dose chemotherapy and immunotherapy with myeloablative or nonmyeloablative allogeneic peripheral blood SCT.[25,26,27,28,29,30] Although most patients who attain complete remission after autologous SCT eventually relapse, a survival plateau for allogeneic SCT suggests an additional graft-versus-leukemia effect.[30] A series (NCT00281983) of 90 patients with relapsed or refractory CLL who underwent allogeneic SCT reported a 6-year OS rate of 58% and a 6-year EFS rate of 38%, which included patients with the worst prognostic factors (such as a TP53 pathogenic variant).[31][Level of evidence C2]
Ofatumumab
Ofatumumab is a humanized anti-CD20 monoclonal antibody.
Evidence (ofatumumab alone and in combination with chlorambucil):
A prospective trial included 474 previously treated patients who attained partial or complete remission to second- or third-line chemotherapy. Patients were randomly assigned to 2 years of maintenance therapy with ofatumumab versus observation.[32]
With a median follow-up of 19 months, median PFS favored the ofatumumab maintenance arm, at 29.4 months versus 15.2 months (HR, 0.50; 95% CI, 0.38–0.66; P < .0001). There was no difference in OS.[32][Level of evidence B1]
A prospective randomized trial of 447 patients who were previously untreated compared ofatumumab plus chlorambucil with chlorambucil alone.[33]
With a median follow-up of 2 years, median PFS favored the ofatumumab plus chlorambucil arm, at 22.4 months versus 13.1 months (HR, 0.57; 95% CI, 0.45–0.72; P = .0001). There was no difference in OS.[33][Level of evidence B1]
Involved-field radiation therapy
Relatively low doses of radiation therapy can be administered for lymphadenopathy that causes problems due to size or encroachment on adjacent organs. Sometimes radiation therapy to one nodal area or the spleen will result in an abscopal effect (i.e., the shrinkage of lymph nodes in untreated sites).
Alemtuzumab, the monoclonal antibody directed at CD52, shows activity in the setting of chemotherapy-resistant disease or high-risk untreated patients with del(17p) or TP53 pathogenic variants.[34,35,36] As a single agent, the subcutaneous route of delivery is preferred to the intravenous route in patients because of the similar efficacy and decreased adverse effects, including less acute allergic reactions that were shown in some nonrandomized reports.[36,37,38,39,40]
In a combination regimen, subcutaneous alemtuzumab plus fludarabine (with or without cyclophosphamide) or intravenous alemtuzumab plus alkylating agents have resulted in excess infectious toxicities and death, with no compensatory improvement in efficacy in three phase II trials and one randomized trial.[41,42,43][Level of evidence C3]; [44][Level of evidence B1]
Current Clinical Trials
Use our advanced clinical trial search to find NCI-supported cancer clinical trials that are now enrolling patients. The search can be narrowed by location of the trial, type of treatment, name of the drug, and other criteria. General information about clinical trials is also available.
References:
Jones JA, Mato AR, Wierda WG, et al.: Venetoclax for chronic lymphocytic leukaemia progressing after ibrutinib: an interim analysis of a multicentre, open-label, phase 2 trial. Lancet Oncol 19 (1): 65-75, 2018.
Coutre S, Choi M, Furman RR, et al.: Venetoclax for patients with chronic lymphocytic leukemia who progressed during or after idelalisib therapy. Blood 131 (15): 1704-1711, 2018.
Kater AP, Wu JQ, Kipps T, et al.: Venetoclax Plus Rituximab in Relapsed Chronic Lymphocytic Leukemia: 4-Year Results and Evaluation of Impact of Genomic Complexity and Gene Mutations From the MURANO Phase III Study. J Clin Oncol 38 (34): 4042-4054, 2020.
Mato AR, Hill BT, Lamanna N, et al.: Optimal sequencing of ibrutinib, idelalisib, and venetoclax in chronic lymphocytic leukemia: results from a multicenter study of 683 patients. Ann Oncol 28 (5): 1050-1056, 2017.
Kater AP, Arslan Ö, Demirkan F, et al.: Activity of venetoclax in patients with relapsed or refractory chronic lymphocytic leukaemia: analysis of the VENICE-1 multicentre, open-label, single-arm, phase 3b trial. Lancet Oncol 25 (4): 463-473, 2024.
Thompson PA, Tam CS: Pirtobrutinib: a new hope for patients with BTK inhibitor-refractory lymphoproliferative disorders. Blood 141 (26): 3137-3142, 2023.
Mato AR, Woyach JA, Brown JR, et al.: Pirtobrutinib after a Covalent BTK Inhibitor in Chronic Lymphocytic Leukemia. N Engl J Med 389 (1): 33-44, 2023.
Wierda WG, Shah NN, Cheah CY, et al.: Pirtobrutinib, a highly selective, non-covalent (reversible) BTK inhibitor in patients with B-cell malignancies: analysis of the Richter transformation subgroup from the multicentre, open-label, phase 1/2 BRUIN study. Lancet Haematol 11 (9): e682-e692, 2024.
Porter DL, Levine BL, Kalos M, et al.: Chimeric antigen receptor-modified T cells in chronic lymphoid leukemia. N Engl J Med 365 (8): 725-33, 2011.
Turtle CJ, Hay KA, Hanafi LA, et al.: Durable Molecular Remissions in Chronic Lymphocytic Leukemia Treated With CD19-Specific Chimeric Antigen Receptor-Modified T Cells After Failure of Ibrutinib. J Clin Oncol 35 (26): 3010-3020, 2017.
Siddiqi T, Soumerai JD, Dorritie KA: Rapid undetectable MRD (uMRD) responses in patients with relapsed/refractory (R/R) chronic lymphocytic leukemia/small lymphocytic lymphoma (CLL/SLL) treated with lisocabtagene maraleucel (liso-cel), a CD19-directed CAR T cell product: updated results from Transcend CLL 004, a phase 1/2 study including patients with high-risk disease previously treated with ibrutinib. [Abstract] Blood 134 (Suppl 1): 503, 2019.
Frey NV, Gill S, Hexner EO, et al.: Long-Term Outcomes From a Randomized Dose Optimization Study of Chimeric Antigen Receptor Modified T Cells in Relapsed Chronic Lymphocytic Leukemia. J Clin Oncol 38 (25): 2862-2871, 2020.
Patel K, Danilov AV, Pagel JM: Duvelisib for CLL/SLL and follicular non-Hodgkin lymphoma. Blood 134 (19): 1573-1577, 2019.
Flinn IW, Hillmen P, Montillo M, et al.: The phase 3 DUO trial: duvelisib vs ofatumumab in relapsed and refractory CLL/SLL. Blood 132 (23): 2446-2455, 2018.
U.S. Food and Drug Administration: FDA warns about possible increased risk of death and serious side effects with cancer drug Copiktra (duvelisib). Silver Spring, Md: U.S. Food and Drug Administration, 2022. Available online. Last accessed April 3, 2025.
Chen CI, Bergsagel PL, Paul H, et al.: Single-agent lenalidomide in the treatment of previously untreated chronic lymphocytic leukemia. J Clin Oncol 29 (9): 1175-81, 2011.
Chanan-Khan A, Miller KC, Musial L, et al.: Clinical efficacy of lenalidomide in patients with relapsed or refractory chronic lymphocytic leukemia: results of a phase II study. J Clin Oncol 24 (34): 5343-9, 2006.
Ferrajoli A, Lee BN, Schlette EJ, et al.: Lenalidomide induces complete and partial remissions in patients with relapsed and refractory chronic lymphocytic leukemia. Blood 111 (11): 5291-7, 2008.
Strati P, Keating MJ, Wierda WG, et al.: Lenalidomide induces long-lasting responses in elderly patients with chronic lymphocytic leukemia. Blood 122 (5): 734-7, 2013.
Wendtner CM, Hillmen P, Mahadevan D, et al.: Final results of a multicenter phase 1 study of lenalidomide in patients with relapsed or refractory chronic lymphocytic leukemia. Leuk Lymphoma 53 (3): 417-23, 2012.
Badoux XC, Keating MJ, Wen S, et al.: Phase II study of lenalidomide and rituximab as salvage therapy for patients with relapsed or refractory chronic lymphocytic leukemia. J Clin Oncol 31 (5): 584-91, 2013.
Takahashi K, Hu B, Wang F, et al.: Clinical implications of cancer gene mutations in patients with chronic lymphocytic leukemia treated with lenalidomide. Blood 131 (16): 1820-1832, 2018.
Sutton L, Chevret S, Tournilhac O, et al.: Autologous stem cell transplantation as a first-line treatment strategy for chronic lymphocytic leukemia: a multicenter, randomized, controlled trial from the SFGM-TC and GFLLC. Blood 117 (23): 6109-19, 2011.
Toze CL, Dalal CB, Nevill TJ, et al.: Allogeneic haematopoietic stem cell transplantation for chronic lymphocytic leukaemia: outcome in a 20-year cohort. Br J Haematol 158 (2): 174-85, 2012.
Khouri IF, Saliba RM, Admirand J, et al.: Graft-versus-leukaemia effect after non-myeloablative haematopoietic transplantation can overcome the unfavourable expression of ZAP-70 in refractory chronic lymphocytic leukaemia. Br J Haematol 137 (4): 355-63, 2007.
Sorror ML, Storer BE, Sandmaier BM, et al.: Five-year follow-up of patients with advanced chronic lymphocytic leukemia treated with allogeneic hematopoietic cell transplantation after nonmyeloablative conditioning. J Clin Oncol 26 (30): 4912-20, 2008.
Schetelig J, van Biezen A, Brand R, et al.: Allogeneic hematopoietic stem-cell transplantation for chronic lymphocytic leukemia with 17p deletion: a retrospective European Group for Blood and Marrow Transplantation analysis. J Clin Oncol 26 (31): 5094-100, 2008.
Malhotra P, Hogan WJ, Litzow MR, et al.: Long-term outcome of allogeneic stem cell transplantation in chronic lymphocytic leukemia: analysis after a minimum follow-up of 5 years. Leuk Lymphoma 49 (9): 1724-30, 2008.
Dreger P, Döhner H, Ritgen M, et al.: Allogeneic stem cell transplantation provides durable disease control in poor-risk chronic lymphocytic leukemia: long-term clinical and MRD results of the German CLL Study Group CLL3X trial. Blood 116 (14): 2438-47, 2010.
Dreger P, Schnaiter A, Zenz T, et al.: TP53, SF3B1, and NOTCH1 mutations and outcome of allotransplantation for chronic lymphocytic leukemia: six-year follow-up of the GCLLSG CLL3X trial. Blood 121 (16): 3284-8, 2013.
van Oers MH, Kuliczkowski K, Smolej L, et al.: Ofatumumab maintenance versus observation in relapsed chronic lymphocytic leukaemia (PROLONG): an open-label, multicentre, randomised phase 3 study. Lancet Oncol 16 (13): 1370-9, 2015.
Hillmen P, Robak T, Janssens A, et al.: Chlorambucil plus ofatumumab versus chlorambucil alone in previously untreated patients with chronic lymphocytic leukaemia (COMPLEMENT 1): a randomised, multicentre, open-label phase 3 trial. Lancet 385 (9980): 1873-83, 2015.
Moreton P, Kennedy B, Lucas G, et al.: Eradication of minimal residual disease in B-cell chronic lymphocytic leukemia after alemtuzumab therapy is associated with prolonged survival. J Clin Oncol 23 (13): 2971-9, 2005.
Parikh SA, Keating MJ, O'Brien S, et al.: Frontline chemoimmunotherapy with fludarabine, cyclophosphamide, alemtuzumab, and rituximab for high-risk chronic lymphocytic leukemia. Blood 118 (8): 2062-8, 2011.
Pettitt AR, Jackson R, Carruthers S, et al.: Alemtuzumab in combination with methylprednisolone is a highly effective induction regimen for patients with chronic lymphocytic leukemia and deletion of TP53: final results of the national cancer research institute CLL206 trial. J Clin Oncol 30 (14): 1647-55, 2012.
Stilgenbauer S, Zenz T, Winkler D, et al.: Subcutaneous alemtuzumab in fludarabine-refractory chronic lymphocytic leukemia: clinical results and prognostic marker analyses from the CLL2H study of the German Chronic Lymphocytic Leukemia Study Group. J Clin Oncol 27 (24): 3994-4001, 2009.
Cortelezzi A, Pasquini MC, Gardellini A, et al.: Low-dose subcutaneous alemtuzumab in refractory chronic lymphocytic leukaemia (CLL): results of a prospective, single-arm multicentre study. Leukemia 23 (11): 2027-33, 2009.
Osterborg A, Foà R, Bezares RF, et al.: Management guidelines for the use of alemtuzumab in chronic lymphocytic leukemia. Leukemia 23 (11): 1980-8, 2009.
Gritti G, Reda G, Maura F, et al.: Low dose alemtuzumab in patients with fludarabine-refractory chronic lymphocytic leukemia. Leuk Lymphoma 53 (3): 424-9, 2012.
Lin TS, Donohue KA, Byrd JC, et al.: Consolidation therapy with subcutaneous alemtuzumab after fludarabine and rituximab induction therapy for previously untreated chronic lymphocytic leukemia: final analysis of CALGB 10101. J Clin Oncol 28 (29): 4500-6, 2010.
Badoux XC, Keating MJ, Wang X, et al.: Cyclophosphamide, fludarabine, alemtuzumab, and rituximab as salvage therapy for heavily pretreated patients with chronic lymphocytic leukemia. Blood 118 (8): 2085-93, 2011.
Lepretre S, Aurran T, Mahé B, et al.: Excess mortality after treatment with fludarabine and cyclophosphamide in combination with alemtuzumab in previously untreated patients with chronic lymphocytic leukemia in a randomized phase 3 trial. Blood 119 (22): 5104-10, 2012.
Geisler CH, van T' Veer MB, Jurlander J, et al.: Frontline low-dose alemtuzumab with fludarabine and cyclophosphamide prolongs progression-free survival in high-risk CLL. Blood 123 (21): 3255-62, 2014.
Key References for Chronic Lymphocytic Leukemia Treatment
These references have been identified by members of the PDQ Adult Treatment Editorial Board as significant in the field of chronic lymphocytic leukemia (CLL) treatment. This list is provided to inform users of important studies that have helped shape the current understanding of and treatment options for CLL. Listed after each reference are the sections within this summary where the reference is cited.
Seymour JF, Kipps TJ, Eichhorst BF, et al.: Four-year analysis of Murano study confirms sustained benefit of time-limited venetoclax-rituximab (VenR) in relapsed/refractory (R/R) chronic lymphocytic leukemia (CLL). [Abstract] Blood 134 (Suppl 1): A-355, 2019.
Shanafelt TD, Wang XV, Kay NE, et al.: Ibrutinib-Rituximab or Chemoimmunotherapy for Chronic Lymphocytic Leukemia. N Engl J Med 381 (5): 432-443, 2019. [PUBMED Abstract]
Shanafelt TD, Wang V, Kay NE, et al.: Ibrutinib and rituximab provides superior clinical outcome compared to FCR in younger patients with chronic lymphocytic leukemia (CLL): extended follow-up from the E1912 trial. [Abstract] Blood 134 (Suppl 1): A-33, 2019.
Al-Sawaf O, Zhang C, Tandon M, et al.: Venetoclax plus obinutuzumab versus chlorambucil plus obinutuzumab for previously untreated chronic lymphocytic leukaemia (CLL14): follow-up results from a multicentre, open-label, randomised, phase 3 trial. Lancet Oncology 21 (9): 1188-1200, 2020. [PUBMED Abstract]
The PDQ cancer information summaries are reviewed regularly and updated as new information becomes available. This section describes the latest changes made to this summary as of the date above.
Revised text about the results of a prospective trial that included 432 previously untreated patients with significant medical comorbidities. Patients were randomly assigned to receive either venetoclax plus obinutuzumab or chlorambucil plus obinutuzumab (cited Al-Sawaf et al. as reference 18).
Added text about a prospective trial that included 867 patients with previously untreated CLL. Patients were randomly assigned to one of three treatment regimens: (1) acalabrutinib and venetoclax, (2) acalabrutinib, venetoclax, and obinutuzumab, or (3) either bendamustine and rituximab or fludarabine, cyclophosphamide, and rituximab (at the discretion of the investigator) (cited Brown et al. as reference 29 and level of evidence A1).
Added text to state that the National Comprehensive Cancer Network has listed the combination of venetoclax and ibrutinib as a first-line treatment option for patients with previously untreated CLL (cited Wierda et al. as reference 42).
Revised text about the results of a prospective phase I/II study of pirtobrutinib in 317 patients with CLL/small lymphocytic leukemia (cited Wierda et al. as reference 8 and level of evidence C3).
This summary is written and maintained by the PDQ Adult Treatment Editorial Board, which is editorially independent of NCI. The summary reflects an independent review of the literature and does not represent a policy statement of NCI or NIH. More information about summary policies and the role of the PDQ Editorial Boards in maintaining the PDQ summaries can be found on the About This PDQ Summary and PDQ® Cancer Information for Health Professionals pages.
About This PDQ Summary
Purpose of This Summary
This PDQ cancer information summary for health professionals provides comprehensive, peer-reviewed, evidence-based information about the treatment of chronic lymphocytic leukemia. It is intended as a resource to inform and assist clinicians in the care of their patients. It does not provide formal guidelines or recommendations for making health care decisions.
Reviewers and Updates
This summary is reviewed regularly and updated as necessary by the PDQ Adult Treatment Editorial Board, which is editorially independent of the National Cancer Institute (NCI). The summary reflects an independent review of the literature and does not represent a policy statement of NCI or the National Institutes of Health (NIH).
Board members review recently published articles each month to determine whether an article should:
be discussed at a meeting,
be cited with text, or
replace or update an existing article that is already cited.
Changes to the summaries are made through a consensus process in which Board members evaluate the strength of the evidence in the published articles and determine how the article should be included in the summary.
The lead reviewer for Chronic Lymphocytic Leukemia Treatment is:
Eric J. Seifter, MD (Johns Hopkins University)
Any comments or questions about the summary content should be submitted to Cancer.gov through the NCI website's Email Us. Do not contact the individual Board Members with questions or comments about the summaries. Board members will not respond to individual inquiries.
Levels of Evidence
Some of the reference citations in this summary are accompanied by a level-of-evidence designation. These designations are intended to help readers assess the strength of the evidence supporting the use of specific interventions or approaches. The PDQ Adult Treatment Editorial Board uses a formal evidence ranking system in developing its level-of-evidence designations.
Permission to Use This Summary
PDQ is a registered trademark. Although the content of PDQ documents can be used freely as text, it cannot be identified as an NCI PDQ cancer information summary unless it is presented in its entirety and is regularly updated. However, an author would be permitted to write a sentence such as "NCI's PDQ cancer information summary about breast cancer prevention states the risks succinctly: [include excerpt from the summary]."
The preferred citation for this PDQ summary is:
PDQ® Adult Treatment Editorial Board. PDQ Chronic Lymphocytic Leukemia Treatment. Bethesda, MD: National Cancer Institute. Updated <MM/DD/YYYY>. Available at: https://www.cancer.gov/types/leukemia/hp/cll-treatment-pdq. Accessed <MM/DD/YYYY>. [PMID: 26389470]
Images in this summary are used with permission of the author(s), artist, and/or publisher for use within the PDQ summaries only. Permission to use images outside the context of PDQ information must be obtained from the owner(s) and cannot be granted by the National Cancer Institute. Information about using the illustrations in this summary, along with many other cancer-related images, is available in Visuals Online, a collection of over 2,000 scientific images.
Disclaimer
Based on the strength of the available evidence, treatment options may be described as either "standard" or "under clinical evaluation." These classifications should not be used as a basis for insurance reimbursement determinations. More information on insurance coverage is available on Cancer.gov on the Managing Cancer Care page.
Contact Us
More information about contacting us or receiving help with the Cancer.gov website can be found on our Contact Us for Help page. Questions can also be submitted to Cancer.gov through the website's Email Us.
This information does not replace the advice of a doctor. Ignite Healthwise, LLC disclaims any warranty or liability for your use of this information. Your use of this information means that you agree to the Terms of Use and Privacy Policy. Learn how we develop our content.
Healthwise, Healthwise for every health decision, and the Healthwise logo are trademarks of Ignite Healthwise, LLC.
The Health Encyclopedia contains general health information. Not all treatments or services described are covered benefits for Kaiser Permanente members or offered as services by Kaiser Permanente. For a list of covered benefits, please refer to your Evidence of Coverage or Summary Plan Description. For recommended treatments, please consult with your health care provider.