This information is produced and provided by the National Cancer Institute (NCI). The information in this topic may have changed since it was written. For the most current information, contact the National Cancer Institute via the Internet web site at http://cancer.gov or call 1-800-4-CANCER.
General Information About Childhood Ependymoma
Primary brain tumors, including ependymomas, are a diverse group of diseases that together constitute the most common solid tumor of childhood. Immunohistochemical analysis, cytogenetic and molecular genetic findings, and measures of mitotic activity are increasingly used in tumor diagnosis and classification. Brain tumors are classified according to histology, but tumor location, extent of spread, molecular features, and age are important factors that affect treatment and prognosis.
According to the 2021 revision to the World Health Organization (WHO) Classification of Tumors of the Central Nervous System (CNS), ependymal tumors are classified into the following ten main subtypes based on anatomical site and histopathological and molecular features:[1,2,3]
Supratentorial ependymoma.
Supratentorial ependymoma, ZFTA fusion–positive (formerly called RELA fusion–positive).
Supratentorial ependymoma, YAP1 fusion–positive.
Posterior fossa ependymoma.
Posterior fossa ependymoma, group PFA.
Posterior fossa ependymoma, group PFB.
Spinal ependymoma.
Spinal ependymoma, MYCN-amplified.
Myxopapillary ependymoma.
Subependymoma (supratentorial, posterior fossa, and spinal locations).
The PDQ childhood brain tumor treatment summaries are organized primarily according to the WHO Classification of Tumors of the CNS.[1,3] For a description of the classification of nervous system tumors and a link to the corresponding treatment summary for each type of brain tumor, see Childhood Brain and Spinal Cord Tumors Summary Index.
Incidence
Childhood ependymoma comprises approximately 9% of all childhood brain and spinal cord tumors, representing about 200 cases per year in the United States.[4,5]
Anatomy
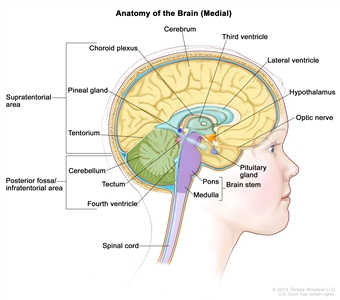
Ependymomas arise from ependymal cells that line the ventricles and passageways in the brain and the center of the spinal cord (see Figure 1). Ependymal cells produce cerebrospinal fluid (CSF). These tumors are classified as supratentorial, posterior fossa (infratentorial), or spinal. In children, 65% to 75% of ependymomas arise in the posterior fossa around the fourth ventricle.[6] Less commonly, ependymomas present in the supratentorial compartment. Spinal ependymomas are rare in childhood.
Figure 1. Anatomy of the inside of the brain, showing the pineal and pituitary glands, optic nerve, ventricles (with cerebrospinal fluid shown in blue), and other parts of the brain. The tentorium separates the cerebrum from the cerebellum. The infratentorium (posterior fossa) is the region below the tentorium that contains the brain stem, cerebellum, and fourth ventricle. The supratentorium is the region above the tentorium and denotes the region that contains the cerebrum.
Clinical Features
The clinical presentation of ependymoma is dependent on tumor location.
Posterior fossa (infratentorial) ependymomas: Children with posterior fossa ependymomas may present with signs and symptoms of obstructive hydrocephalus caused by obstruction at the level of the fourth ventricle. They may also present with ataxia, neck pain, and/or cranial nerve palsies.
Supratentorial ependymomas: Supratentorial ependymomas may result in headaches, seizures, or location-dependent focal neurological deficits.
Spinal cord ependymomas: Spinal cord ependymomas, which are often the myxopapillary variant, tend to cause back pain, lower extremity weakness, and/or bowel and bladder dysfunction.
Diagnostic Evaluation
Every patient suspected of having an ependymoma is evaluated with diagnostic imaging of the whole brain and spinal cord. The most sensitive method available for evaluating spinal cord subarachnoid metastasis is spinal magnetic resonance imaging (MRI) performed with gadolinium. This is ideally done before surgery to avoid confusion with postoperative blood. If MRI is used, the entire spine is generally imaged in at least two planes with contiguous MRI slices performed after gadolinium enhancement.
If feasible, CSF cytological evaluation is conducted.[7] Despite the frequent finding of disseminated disease at the time of recurrence, metastatic disease at initial presentation is rare.[8][Level of evidence C2]
Prognostic Factors
Unfavorable factors affecting outcome (except as noted) include the following:
Molecular characteristics.
Posterior fossa ependymomas are divided into the following two primary molecular groups on the basis of distinctive patterns of gene expression.[9,10,11,12]
Posterior fossa A ependymoma (PF-EPN-A).
PF-EPN-A occurs primarily in young children and is characterized by a largely balanced genomic profile, with an increased occurrence of chromosome 1q gain [13,14,15,16] and expression of genes and proteins previously shown to be associated with poor prognosis, such as tenascin C and epidermal growth factor receptor.[13,17,18]
Gain of 1q confers a very poor prognosis despite complete resection and postoperative radiation therapy (5-year event-free survival rate, 81.5% for balanced 1q vs. 35.7% for gain 1q).[19][Level of evidence B4]
A combined retrospective analysis of 663 patients from five nonoverlapping cohorts identified loss of 6q as a poor prognostic factor for patients with PF-EPN-A.[20] Loss of 6q was observed in 8.6% of PF-EPN-A cases, and it is more common in tumors with 1q gain. The subset of patients (n = 22) with both 1q gain and 6q loss had a particularly poor prognosis.
A retrospective multi-institutional study compared patient-matched primary tumors with recurrent tumors. The study reported that the high-risk features of 1q gain and 6q loss were more frequent in recurrent tumors than in primary tumors, and these features remained associated with a poor prognosis.[21]
Posterior fossa B ependymoma (PF-EPN-B).
PF-EPN-B occurs primarily in older children and adults and is characterized by a more favorable prognosis and by numerous cytogenetic abnormalities involving whole chromosomes or chromosomal arms.[9,12,22]
Patients with PF-EPN-B have a favorable outcome when compared with patients with PF-EPN-A. Patients with PF-EPN-B have a 5-year progression-free survival (PFS) rate of 73% and an overall survival (OS) rate exceeding 90%.[11,12]
Gain of 1q is not a prognostic feature in patients with PF-EPN-B, whereas loss of chromosome 13q may confer a poor prognosis.[22]
Supratentorial ependymomas can be divided into the following two primary molecular groups on the basis of their gene fusion status:
While a retrospective analysis suggested that the RELA fusion predicted poorer prognosis,[11] subsequent reports suggest that patients with RELA fusions who undergo a complete resection and postoperative radiation have relatively favorable survival rates that are in the range of 80% at 5 years.[11,19,23,24] Retrospective studies suggest a poor outcome for patients who undergo complete surgical resections but do not receive postoperative radiation therapy.[11]
Homozygous deletion of CDKN2A has been associated with a poor prognosis in patients with ST-EPN-ZFTA.[25][Level of evidence B4] CDKN2A deletion has also been reported as a secondary event in recurrent ependymoma.[26]
Supratentorial ependymoma with YAP1 fusions (ST-EPN-YAP1).
Patients with ST-EPN-YAP1 have a favorable prognosis (although based on small numbers), with 5-year survival rates approaching 100%.[11,23,27]
Spinal ependymomas can be separated by methylome studies, but molecular classification does not provide any clinicopathological advantage over histopathological classification for myxopapillary ependymoma and subependymoma. However, molecular classification is useful for identifying spinal ependymoma with MYCN amplification, which has been associated with a poor prognosis. There is a paucity of data on the optimal risk stratification of spinal ependymoma in children, although inferring from adults, a complete resection confers a favorable prognosis.
Spinal ependymoma, MYCN-amplified (SP-EPN-MYCN).
This is a rare and aggressive ependymoma that predominantly affects young adults.
SP-EPN-MYCN tumors are typically grade 3, and they are characterized by aggressive behavior, with frequent leptomeningeal dissemination and high rate of recurrence.[28,29,30,31]
Younger age at diagnosis. Younger age at diagnosis has historically been a poor prognostic factor, although this could partially result from the common practice of avoiding or deferring radiation in children younger than 3 years. In a prospective Children's Oncology Group (COG) trial (ACNS0121 [NCT00027846]), immediate postoperative radiation therapy was given to all children older than 1 year after gross-total resection or near-total resection. The study demonstrated that there was no significant difference in 5-year PFS or OS between patients aged 1 to 3 years and patients aged 3 to 21 years.[19]
Anaplastic histology. Anaplastic histology has been associated with a poor prognosis.[32][Level of evidence B4]; [33,34,35,36]; [37][Level of evidence C1]; [38][Level of evidence C2] However, the distinction between grade 2 and grade 3 disease has significant interobserver variability, confounding the use of anaplasia as a prognostic factor.[39] The 2021 WHO Classification of Tumors of the CNS no longer uses the term anaplastic ependymoma and allows only a histologically defined diagnosis of ependymoma in the integrated diagnosis. Within the layered report, a pathologist can still choose to assign either CNS WHO grade 2 or 3 to a tumor on the basis of its histological features.[2,3]
Subtotal resection. Subtotal resection confers a very poor prognosis.[19,35,36]; [32][Level of evidence B4]
Lower doses of radiation. Lower doses of radiation or chemotherapy-only protocols confer a poor prognosis.[12,23,40,41]
Follow-Up After Treatment
Surveillance neuroimaging, coupled with clinical assessments, is generally recommended after treatment for ependymoma. In a report of 198 patients with ependymoma, 90 experienced a relapse. Patients whose relapsed tumor was detected by routine surveillance imaging had superior second PFS than patients whose relapsed tumor was detected by clinical symptomology. The latter were more likely to have metastatic disease at relapse. It is not known whether these patients also had more biologically aggressive disease, although the median time to relapse and the median time from last surveillance imaging was the same in both groups.[42]
Most practitioners obtain MRI of the brain and/or spinal cord at the following intervals:[43][Level of evidence B4]
First 2 to 3 years after treatment: Every 3 to 4 months.
Four to 5 years after treatment: Every 6 months.
More than 5 years after treatment: Annually because of the high incidence of late recurrences.
References:
Louis DN, Ohgaki H, Wiestler OD: WHO Classification of Tumours of the Central Nervous System. 4th rev.ed. IARC Press, 2016.
Louis DN, Perry A, Wesseling P, et al.: The 2021 WHO Classification of Tumors of the Central Nervous System: a summary. Neuro Oncol 23 (8): 1231-1251, 2021.
WHO Classification of Tumours Editorial Board, ed.: WHO Classification of Tumours: Central Nervous System Tumours. Vol. 6. 5th ed. IARC Press; 2021.
Gurney JG, Smith MA, Bunin GR: CNS and miscellaneous intracranial and intraspinal neoplasms. In: Ries LA, Smith MA, Gurney JG, et al., eds.: Cancer incidence and survival among children and adolescents: United States SEER Program 1975-1995. National Cancer Institute, SEER Program, 1999. NIH Pub.No. 99-4649, Chapter 3, pp 51-63. Also available online. Last accessed February 9, 2024.
Ostrom QT, Gittleman H, Truitt G, et al.: CBTRUS Statistical Report: Primary Brain and Other Central Nervous System Tumors Diagnosed in the United States in 2011-2015. Neuro Oncol 20 (suppl_4): iv1-iv86, 2018.
Andreiuolo F, Puget S, Peyre M, et al.: Neuronal differentiation distinguishes supratentorial and infratentorial childhood ependymomas. Neuro Oncol 12 (11): 1126-34, 2010.
Moreno L, Pollack IF, Duffner PK, et al.: Utility of cerebrospinal fluid cytology in newly diagnosed childhood ependymoma. J Pediatr Hematol Oncol 32 (6): 515-8, 2010.
Benesch M, Mynarek M, Witt H, et al.: Newly Diagnosed Metastatic Intracranial Ependymoma in Children: Frequency, Molecular Characteristics, Treatment, and Outcome in the Prospective HIT Series. Oncologist 24 (9): e921-e929, 2019.
Wani K, Armstrong TS, Vera-Bolanos E, et al.: A prognostic gene expression signature in infratentorial ependymoma. Acta Neuropathol 123 (5): 727-38, 2012.
Witt H, Mack SC, Ryzhova M, et al.: Delineation of two clinically and molecularly distinct subgroups of posterior fossa ependymoma. Cancer Cell 20 (2): 143-57, 2011.
Pajtler KW, Witt H, Sill M, et al.: Molecular Classification of Ependymal Tumors across All CNS Compartments, Histopathological Grades, and Age Groups. Cancer Cell 27 (5): 728-43, 2015.
Ramaswamy V, Hielscher T, Mack SC, et al.: Therapeutic Impact of Cytoreductive Surgery and Irradiation of Posterior Fossa Ependymoma in the Molecular Era: A Retrospective Multicohort Analysis. J Clin Oncol 34 (21): 2468-77, 2016.
Mendrzyk F, Korshunov A, Benner A, et al.: Identification of gains on 1q and epidermal growth factor receptor overexpression as independent prognostic markers in intracranial ependymoma. Clin Cancer Res 12 (7 Pt 1): 2070-9, 2006.
Korshunov A, Witt H, Hielscher T, et al.: Molecular staging of intracranial ependymoma in children and adults. J Clin Oncol 28 (19): 3182-90, 2010.
Kilday JP, Mitra B, Domerg C, et al.: Copy number gain of 1q25 predicts poor progression-free survival for pediatric intracranial ependymomas and enables patient risk stratification: a prospective European clinical trial cohort analysis on behalf of the Children's Cancer Leukaemia Group (CCLG), Societe Francaise d'Oncologie Pediatrique (SFOP), and International Society for Pediatric Oncology (SIOP). Clin Cancer Res 18 (7): 2001-11, 2012.
Godfraind C, Kaczmarska JM, Kocak M, et al.: Distinct disease-risk groups in pediatric supratentorial and posterior fossa ependymomas. Acta Neuropathol 124 (2): 247-57, 2012.
Korshunov A, Golanov A, Timirgaz V: Immunohistochemical markers for intracranial ependymoma recurrence. An analysis of 88 cases. J Neurol Sci 177 (1): 72-82, 2000.
Andreiuolo F, Le Teuff G, Bayar MA, et al.: Integrating Tenascin-C protein expression and 1q25 copy number status in pediatric intracranial ependymoma prognostication: A new model for risk stratification. PLoS One 12 (6): e0178351, 2017.
Merchant TE, Bendel AE, Sabin ND, et al.: Conformal Radiation Therapy for Pediatric Ependymoma, Chemotherapy for Incompletely Resected Ependymoma, and Observation for Completely Resected, Supratentorial Ependymoma. J Clin Oncol 37 (12): 974-983, 2019.
Baroni LV, Sundaresan L, Heled A, et al.: Ultra high-risk PFA ependymoma is characterized by loss of chromosome 6q. Neuro Oncol 23 (8): 1360-1370, 2021.
Donson AM, Bertrand KC, Riemondy KA, et al.: Significant increase of high-risk chromosome 1q gain and 6q loss at recurrence in posterior fossa group A ependymoma: A multicenter study. Neuro Oncol 25 (10): 1854-1867, 2023.
Cavalli FMG, Hübner JM, Sharma T, et al.: Heterogeneity within the PF-EPN-B ependymoma subgroup. Acta Neuropathol 136 (2): 227-237, 2018.
Upadhyaya SA, Robinson GW, Onar-Thomas A, et al.: Molecular grouping and outcomes of young children with newly diagnosed ependymoma treated on the multi-institutional SJYC07 trial. Neuro Oncol 21 (10): 1319-1330, 2019.
Fukuoka K, Kanemura Y, Shofuda T, et al.: Significance of molecular classification of ependymomas: C11orf95-RELA fusion-negative supratentorial ependymomas are a heterogeneous group of tumors. Acta Neuropathol Commun 6 (1): 134, 2018.
Jünger ST, Andreiuolo F, Mynarek M, et al.: CDKN2A deletion in supratentorial ependymoma with RELA alteration indicates a dismal prognosis: a retrospective analysis of the HIT ependymoma trial cohort. Acta Neuropathol 140 (3): 405-407, 2020.
Milde T, Pfister S, Korshunov A, et al.: Stepwise accumulation of distinct genomic aberrations in a patient with progressively metastasizing ependymoma. Genes Chromosomes Cancer 48 (3): 229-38, 2009.
Andreiuolo F, Varlet P, Tauziède-Espariat A, et al.: Childhood supratentorial ependymomas with YAP1-MAMLD1 fusion: an entity with characteristic clinical, radiological, cytogenetic and histopathological features. Brain Pathol 29 (2): 205-216, 2019.
Ghasemi DR, Sill M, Okonechnikov K, et al.: MYCN amplification drives an aggressive form of spinal ependymoma. Acta Neuropathol 138 (6): 1075-1089, 2019.
Swanson AA, Raghunathan A, Jenkins RB, et al.: Spinal Cord Ependymomas With MYCN Amplification Show Aggressive Clinical Behavior. J Neuropathol Exp Neurol 78 (9): 791-797, 2019.
Scheil S, Brüderlein S, Eicker M, et al.: Low frequency of chromosomal imbalances in anaplastic ependymomas as detected by comparative genomic hybridization. Brain Pathol 11 (2): 133-43, 2001.
Raffeld M, Abdullaev Z, Pack SD, et al.: High level MYCN amplification and distinct methylation signature define an aggressive subtype of spinal cord ependymoma. Acta Neuropathol Commun 8 (1): 101, 2020.
Massimino M, Miceli R, Giangaspero F, et al.: Final results of the second prospective AIEOP protocol for pediatric intracranial ependymoma. Neuro Oncol 18 (10): 1451-60, 2016.
Merchant TE, Jenkins JJ, Burger PC, et al.: Influence of tumor grade on time to progression after irradiation for localized ependymoma in children. Int J Radiat Oncol Biol Phys 53 (1): 52-7, 2002.
Korshunov A, Golanov A, Sycheva R, et al.: The histologic grade is a main prognostic factor for patients with intracranial ependymomas treated in the microneurosurgical era: an analysis of 258 patients. Cancer 100 (6): 1230-7, 2004.
Tamburrini G, D'Ercole M, Pettorini BL, et al.: Survival following treatment for intracranial ependymoma: a review. Childs Nerv Syst 25 (10): 1303-12, 2009.
Massimino M, Barretta F, Modena P, et al.: Second series by the Italian Association of Pediatric Hematology and Oncology of children and adolescents with intracranial ependymoma: an integrated molecular and clinical characterization with a long-term follow-up. Neuro Oncol 23 (5): 848-857, 2021.
Amirian ES, Armstrong TS, Aldape KD, et al.: Predictors of survival among pediatric and adult ependymoma cases: a study using Surveillance, Epidemiology, and End Results data from 1973 to 2007. Neuroepidemiology 39 (2): 116-24, 2012.
Tihan T, Zhou T, Holmes E, et al.: The prognostic value of histological grading of posterior fossa ependymomas in children: a Children's Oncology Group study and a review of prognostic factors. Mod Pathol 21 (2): 165-77, 2008.
Ellison DW, Kocak M, Figarella-Branger D, et al.: Histopathological grading of pediatric ependymoma: reproducibility and clinical relevance in European trial cohorts. J Negat Results Biomed 10: 7, 2011.
Vaidya K, Smee R, Williams JR: Prognostic factors and treatment options for paediatric ependymomas. J Clin Neurosci 19 (9): 1228-35, 2012.
Zapotocky M, Beera K, Adamski J, et al.: Survival and functional outcomes of molecularly defined childhood posterior fossa ependymoma: Cure at a cost. Cancer 125 (11): 1867-1876, 2019.
Klawinski D, Indelicato DJ, Hossain J, et al.: Surveillance imaging in pediatric ependymoma. Pediatr Blood Cancer 67 (11): e28622, 2020.
Massimino M, Barretta F, Modena P, et al.: Pediatric intracranial ependymoma: correlating signs and symptoms at recurrence with outcome in the second prospective AIEOP protocol follow-up. J Neurooncol 140 (2): 457-465, 2018.
Molecular Features of Childhood Ependymoma
Molecular Subgroups of Ependymoma
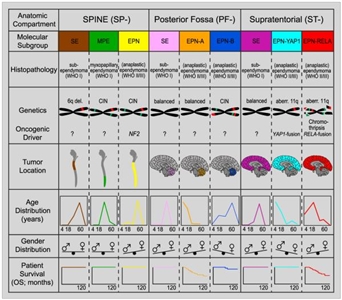
Molecular characterization studies have previously identified nine molecular subgroups of ependymoma, six of which predominate in childhood. The subgroups are determined by their distinctive DNA methylation and gene expression profiles and unique spectrum of genomic alterations (see Figure 2).[1,2,3,4]
One new molecularly defined ependymoma was added to the 2021 World Health Organization (WHO) Classification of Tumours of the Central Nervous System: spinal ependymoma with MYCN amplification. The 2021 classification further described ependymal tumors defined by anatomical location and histology but not by molecular alteration. These tumors are called posterior fossa ependymoma (PF-EPN), supratentorial ependymoma (ST-EPN), and spinal ependymoma (SP-EPN). These tumors either contain a unique molecular alteration (not elsewhere classified [NEC]) or their molecular analysis failed or was not obtained (not otherwise specified [NOS]).[5]
Infratentorial tumors.
Posterior fossa ependymoma (PF-EPN).
Posterior fossa A (PF-EPN-A), loss of H3 K27 trimethylation mark.
Posterior fossa B (PF-EPN-B), retained H3 K27 trimethylation mark.
Supratentorial tumors.
Supratentorial ependymoma (ST-EPN).
ZFTA fusion–positive ependymoma (ST-EPN-ZFTA). This was previously called RELA fusion–positive ependymoma (ST-EPN-RELA), but it was renamed because ZFTA is the new designation for C11orf95, and ZFTA may be fused with a partner gene other than RELA.[6]
YAP1 fusion–positive ependymoma (ST-EPN-YAP1).
Spinal tumors.
Spinal ependymoma (SP-EPN).
Spinal ependymoma, MYCN-amplified (SP-EPN-MYCN).
Myxopapillary ependymoma (SP-EPN-MPE).
Subependymoma—whether supratentorial, infratentorial, or spinal—accounts for the remaining three molecular variants, and it is rarely, if ever, seen in children.
Figure 2. Graphical summary of key molecular and clinical characteristics of ependymal tumor subgroups. Schematic representation of key genetic and epigenetic findings in the nine molecular subgroups of ependymal tumors as identified by methylation profiling. CIN, Chromosomal instability. Reprinted from Cancer Cell, Volume 27, Kristian W. Pajtler, Hendrik Witt, Martin Sill, David T.W. Jones, Volker Hovestadt, Fabian Kratochwil, Khalida Wani, Ruth Tatevossian, Chandanamali Punchihewa, Pascal Johann, Juri Reimand, Hans-Jorg Warnatz, Marina Ryzhova, Steve Mack, Vijay Ramaswamy, David Capper, Leonille Schweizer, Laura Sieber, Andrea Wittmann, Zhiqin Huang, Peter van Sluis, Richard Volckmann, Jan Koster, Rogier Versteeg, Daniel Fults, Helen Toledano, Smadar Avigad, Lindsey M. Hoffman, Andrew M. Donson, Nicholas Foreman, Ekkehard Hewer, Karel Zitterbart, Mark Gilbert, Terri S. Armstrong, Nalin Gupta, Jeffrey C. Allen, Matthias A. Karajannis, David Zagzag, Martin Hasselblatt, Andreas E. Kulozik, Olaf Witt, V. Peter Collins, Katja von Hoff, Stefan Rutkowski, Torsten Pietsch, Gary Bader, Marie-Laure Yaspo, Andreas von Deimling, Peter Lichter, Michael D. Taylor, Richard Gilbertson, David W. Ellison, Kenneth Aldape, Andrey Korshunov, Marcel Kool, and Stefan M. Pfister, Molecular Classification of Ependymal Tumors across All CNS Compartments, Histopathological Grades, and Age Groups, Pages 728–743, Copyright (2015), with permission from Elsevier.
Infratentorial tumors
Posterior fossa A ependymoma (PF-EPN-A)
The most common posterior fossa ependymoma subgroup is PF-EPN-A and is characterized by the following:
Presentation in young children (median age, 3 years).[1,7]
Low rates of variants that affect protein structure, approximately five per genome.[2]
Gain of chromosome 1q, a known poor prognostic factor for patients with ependymoma,[8] in approximately 25% of cases.[1,3,9]
Loss of chromosome 6q, reported to be a poor prognostic factor for patients with PF-EPN-A, in 8% to 10% of cases.[10]
A balanced chromosomal profile with few chromosomal gains or losses.[1,2]
Loss of the H3 K27 trimethylation mark and globally hypomethylated DNA.[11] A prospective multi-institutional study analyzed 147 patients with ependymoma. The study reported high sensitivity and specificity for immunohistochemical detection of loss of the H3 K27 trimethylation mark in identifying PF-EPN-A cases.[12] Loss of this mark occurs through multiple mechanisms, including the following:
Recurrent variants of EZHIP in 10% of cases, with high EZHIP mRNA expression across almost all PF-EPN-A.[13,14] EZHIP expression (with or without alteration) results in inhibition of the methyltransferase EZH2 leading to loss of the H3 K27 trimethylation mark.[14,15]
Recurrent K27M variants in histone H3 variants in a small proportion of cases.[16,17] Unlike diffuse midline gliomas, variants in H3.1 (H3C2 and H3C3) are more common than variants in H3.3 (H3-3A).[13] Histone variants are mutually exclusive with high expression of EZHIP,[13] and they also lead to loss of the H3 K27 trimethylation mark through EZH2 inhibition.
A study that included over 600 cases of PF-EPN-A used methylation array profiling to divide this population into two distinctive subgroups, PFA-1 and PFA-2.[13] Gene expression profiling suggested that these two subtypes may arise in different anatomical locations in the hindbrain. Within both PFA-1 and PFA-2 groups, distinctive minor subtypes could be identified, suggesting the presence of heterogeneity. Additional study will be required to define the clinical significance of these subtypes.
Posterior fossa B ependymoma (PF-EPN-B)
The PF-EPN-B subgroup is less common than the PF-EPN-A subgroup, representing 15% to 20% of all posterior fossa ependymomas in children. PF-EPN-B is characterized by the following:
Presentation primarily in adolescents and young adults (median age, 30 years).[1,7]
Low rates of variants that affect protein structure (approximately five per genome), with no recurring variants.[3]
Numerous cytogenetic abnormalities, primarily involving the gain/loss of whole chromosomes.[1,3]
ST-EPN-ZFTA is the largest subset of pediatric supratentorial ependymomas and is predominantly characterized by gene fusions involving RELA,[19,20] a transcriptional factor important in NF-κB pathway activity. ST-EPN-ZFTA is characterized by the following:
Represents approximately 70% of supratentorial ependymomas in children,[19,20] and presents at a median age of 8 years.[1]
Presence of ZFTA fusions result from chromothripsis involving chromosome 11q13.1.[19]
Low rates of variants that affect protein structure and near absence of recurring variants outside of ZFTA::RELA fusions.[19]
Evidence of NF-κB pathway activation at the protein and RNA level.[19]
Gain of chromosome 1q, in approximately one-quarter of cases, with an indeterminate effect on survival.[1]
The concordance was high between immunohistochemistry for nuclear p65-RelA, fluorescence in situ hybridization for ZFTA and RELA, and DNA methylation-based classification for defining ST-EPN-ZFTA.[21]
Homozygous deletion of CDKN2A has been associated with a poor prognosis in patients with ZFTA fusion–positive ependymoma.[22][Level of evidence B4] CDKN2A deletion has also been reported as a secondary event in recurrent ependymoma.[23]
ST-EPN-YAP1 is the second, less common subset of supratentorial ependymomas and has fusions involving YAP1 on chromosome 11. ST-EPN-YAP1 is characterized by the following:
Presence of a gene fusion involving YAP1, with MAMLD1 being the most common fusion partner.[1,19]
A relatively stable genome with few chromosomal changes other than the YAP1 fusion.[1]
Tumors mimicking supratentorial ependymomas
Supratentorial ependymomas without ZFTA or YAP1 fusions (on chromosome 11) are an undefined entity, and it is unclear what these samples represent. By DNA methylation analysis, these samples often cluster with other entities such as high-grade gliomas and embryonal tumors. As one example, a retrospective methylation analysis of supratentorial brain tumors identified a group of tumors distinct from supratentorial ependymoma that harbor recurrent PLAGL1 fusions.[24] The histological lineage of these PLAGL1-altered tumors is not yet clear. Nineteen of the 32 tumors (59%) had previously been reported as ependymomas. Caution should be taken when diagnosing a supratentorial ependymoma that does not harbor a fusion involving chromosome 11.[6,25,26]
SP-EPN-MYCN is rare, with only 27 cases reported.[27,28,29,30]
Median age at presentation was 31 years (range, 12–56 years).
High level of MYCN amplification was present at diagnosis and relapse.
SP-EPN-MYCN has a unique methylation profile compared with other spinal cord ependymomas, MYCN-amplified pediatric-type glioblastoma, and neuroblastoma.
References:
Pajtler KW, Witt H, Sill M, et al.: Molecular Classification of Ependymal Tumors across All CNS Compartments, Histopathological Grades, and Age Groups. Cancer Cell 27 (5): 728-43, 2015.
Witt H, Mack SC, Ryzhova M, et al.: Delineation of two clinically and molecularly distinct subgroups of posterior fossa ependymoma. Cancer Cell 20 (2): 143-57, 2011.
Pajtler KW, Mack SC, Ramaswamy V, et al.: The current consensus on the clinical management of intracranial ependymoma and its distinct molecular variants. Acta Neuropathol 133 (1): 5-12, 2017.
WHO Classification of Tumours Editorial Board, ed.: WHO Classification of Tumours: Central Nervous System Tumours. Vol. 6. 5th ed. IARC Press; 2021.
Zschernack V, Jünger ST, Mynarek M, et al.: Supratentorial ependymoma in childhood: more than just RELA or YAP. Acta Neuropathol 141 (3): 455-466, 2021.
Ramaswamy V, Hielscher T, Mack SC, et al.: Therapeutic Impact of Cytoreductive Surgery and Irradiation of Posterior Fossa Ependymoma in the Molecular Era: A Retrospective Multicohort Analysis. J Clin Oncol 34 (21): 2468-77, 2016.
Korshunov A, Witt H, Hielscher T, et al.: Molecular staging of intracranial ependymoma in children and adults. J Clin Oncol 28 (19): 3182-90, 2010.
Merchant TE, Bendel AE, Sabin ND, et al.: Conformal Radiation Therapy for Pediatric Ependymoma, Chemotherapy for Incompletely Resected Ependymoma, and Observation for Completely Resected, Supratentorial Ependymoma. J Clin Oncol 37 (12): 974-983, 2019.
Baroni LV, Sundaresan L, Heled A, et al.: Ultra high-risk PFA ependymoma is characterized by loss of chromosome 6q. Neuro Oncol 23 (8): 1360-1370, 2021.
Panwalkar P, Clark J, Ramaswamy V, et al.: Immunohistochemical analysis of H3K27me3 demonstrates global reduction in group-A childhood posterior fossa ependymoma and is a powerful predictor of outcome. Acta Neuropathol 134 (5): 705-714, 2017.
Chapman RJ, Ghasemi DR, Andreiuolo F, et al.: Optimizing biomarkers for accurate ependymoma diagnosis, prognostication, and stratification within International Clinical Trials: A BIOMECA study. Neuro Oncol 25 (10): 1871-1882, 2023.
Pajtler KW, Wen J, Sill M, et al.: Molecular heterogeneity and CXorf67 alterations in posterior fossa group A (PFA) ependymomas. Acta Neuropathol 136 (2): 211-226, 2018.
Hübner JM, Müller T, Papageorgiou DN, et al.: EZHIP/CXorf67 mimics K27M mutated oncohistones and functions as an intrinsic inhibitor of PRC2 function in aggressive posterior fossa ependymoma. Neuro Oncol 21 (7): 878-889, 2019.
Jain SU, Do TJ, Lund PJ, et al.: PFA ependymoma-associated protein EZHIP inhibits PRC2 activity through a H3 K27M-like mechanism. Nat Commun 10 (1): 2146, 2019.
Gessi M, Capper D, Sahm F, et al.: Evidence of H3 K27M mutations in posterior fossa ependymomas. Acta Neuropathol 132 (4): 635-7, 2016.
Ryall S, Guzman M, Elbabaa SK, et al.: H3 K27M mutations are extremely rare in posterior fossa group A ependymoma. Childs Nerv Syst 33 (7): 1047-1051, 2017.
Cavalli FMG, Hübner JM, Sharma T, et al.: Heterogeneity within the PF-EPN-B ependymoma subgroup. Acta Neuropathol 136 (2): 227-237, 2018.
Parker M, Mohankumar KM, Punchihewa C, et al.: C11orf95-RELA fusions drive oncogenic NF-κB signalling in ependymoma. Nature 506 (7489): 451-5, 2014.
Pietsch T, Wohlers I, Goschzik T, et al.: Supratentorial ependymomas of childhood carry C11orf95-RELA fusions leading to pathological activation of the NF-κB signaling pathway. Acta Neuropathol 127 (4): 609-11, 2014.
Pagès M, Pajtler KW, Puget S, et al.: Diagnostics of pediatric supratentorial RELA ependymomas: integration of information from histopathology, genetics, DNA methylation and imaging. Brain Pathol 29 (3): 325-335, 2019.
Jünger ST, Andreiuolo F, Mynarek M, et al.: CDKN2A deletion in supratentorial ependymoma with RELA alteration indicates a dismal prognosis: a retrospective analysis of the HIT ependymoma trial cohort. Acta Neuropathol 140 (3): 405-407, 2020.
Milde T, Pfister S, Korshunov A, et al.: Stepwise accumulation of distinct genomic aberrations in a patient with progressively metastasizing ependymoma. Genes Chromosomes Cancer 48 (3): 229-38, 2009.
Sievers P, Henneken SC, Blume C, et al.: Recurrent fusions in PLAGL1 define a distinct subset of pediatric-type supratentorial neuroepithelial tumors. Acta Neuropathol 142 (5): 827-839, 2021.
Sturm D, Orr BA, Toprak UH, et al.: New Brain Tumor Entities Emerge from Molecular Classification of CNS-PNETs. Cell 164 (5): 1060-72, 2016.
Fukuoka K, Kanemura Y, Shofuda T, et al.: Significance of molecular classification of ependymomas: C11orf95-RELA fusion-negative supratentorial ependymomas are a heterogeneous group of tumors. Acta Neuropathol Commun 6 (1): 134, 2018.
Ghasemi DR, Sill M, Okonechnikov K, et al.: MYCN amplification drives an aggressive form of spinal ependymoma. Acta Neuropathol 138 (6): 1075-1089, 2019.
Swanson AA, Raghunathan A, Jenkins RB, et al.: Spinal Cord Ependymomas With MYCN Amplification Show Aggressive Clinical Behavior. J Neuropathol Exp Neurol 78 (9): 791-797, 2019.
Scheil S, Brüderlein S, Eicker M, et al.: Low frequency of chromosomal imbalances in anaplastic ependymomas as detected by comparative genomic hybridization. Brain Pathol 11 (2): 133-43, 2001.
Raffeld M, Abdullaev Z, Pack SD, et al.: High level MYCN amplification and distinct methylation signature define an aggressive subtype of spinal cord ependymoma. Acta Neuropathol Commun 8 (1): 101, 2020.
Histopathological Classification of Childhood Ependymal Tumors
For the first time, the 2016 World Health Organization (WHO) Classification of Tumours of the Central Nervous System (CNS) incorporated genotypic findings into the classification of select CNS tumors. This integrated classification is intended to define more homogeneous entities that will improve the accuracy of diagnoses, refine prognoses, and more reliably reach conclusions regarding treatment strategies.
The 2021 WHO classification continues to classify ependymal tumors on the basis of anatomical site (i.e., supratentorial, posterior fossa, spinal), histopathological features (i.e., subependymoma, myxopapillary ependymoma, ependymoma), and molecular features (i.e., supratentorial ependymoma with ZFTA [formerly called C11orf95] or YAP1 fusions, posterior fossa A or B, and spinal ependymoma with MYCN amplification). The updated classification also includes ependymal tumors defined by anatomical location and histology but not by molecular alteration. Examples include cases where the tumor contains a unique molecular alteration (in such cases, the term not elsewhere classified [NEC] is used) or when molecular analysis fails or is not feasible (in these cases, the term not otherwise specified [NOS] is used).[1]
Ependymal tumors are now classified into the following three main histological subtypes:[1,2]
Subependymoma (WHO grade 1): A subependymoma is a slow-growing neoplasm, typically attached to the ventricle wall. It is composed of glial tumor cell clusters embedded in a fibrillary matrix.
The true incidence of subependymomas (WHO grade 1) is difficult to determine. These tumors are frequently asymptomatic and may be found incidentally at autopsy. Subependymomas probably comprise less than 5% of all ependymal tumors.
A diagnosis of subependymoma in a child is questionable, and further review or molecular analysis should be considered.[3]
Myxopapillary ependymoma (WHO grade 2): A myxopapillary ependymoma arises almost exclusively in the location of the conus medullaris, cauda equina, and filum terminale of the spinal cord. They are characterized histologically by tumor cells arranged in a papillary manner around vascularized myxoid stromal cores. Myxopapillary ependymoma is now considered WHO grade 2, rather than grade 1, because its recurrence rate is similar to conventional spinal ependymoma.[4]
Ependymoma: Ependymoma originates from the walls of the ventricles or from the spinal canal and are composed of neoplastic ependymal cells.
In the 2016 WHO revision, anaplastic ependymoma was eliminated as a subtype. In the 2021 WHO revision, papillary, clear cell, and tanycytic ependymoma were removed as subtypes because they were of no clinicopathologic utility. They are now included as patterns when describing the histopathology of an ependymoma.
Grading of ependymoma has been fraught with issues of reproducibility and clinical usefulness, especially in molecularly defined ependymoma. Therefore, the 2021 WHO classification allows only a histologically defined diagnosis of ependymoma in the integrated diagnosis (i.e., anaplastic ependymoma is no longer allowed), but a pathologist can choose to assign WHO grade 2 or 3 on the basis of the histopathological features. Grade 3 ependymoma, compared with grade 2 ependymoma, shows increased cellularity and mitotic activity, often associated with microvascular proliferation and necrosis. The distinction between grade 2 and grade 3 has significant interobserver variability and lacks uniformity across cooperative group studies.[5]
Histologically defined ependymoma can be further classified by molecular features, as follows:
Supratentorial ependymoma includes the molecular subtypes ST-EPN (NEC or NOS), ST-EPN-ZFTA, and ST-EPN-YAP1.
Posterior fossa ependymoma includes PF-EPN (NEC or NOS), PF-EPN-A, and PF-EPN-B.
Spinal ependymoma includes SP-EPN (NEC or NOS) and SP-EPN-MYCN.
Subependymoma and myxopapillary ependymoma are usually considered to be clinically and pathologically distinct from spinal ependymoma.
Although supratentorial and infratentorial ependymoma are believed to arise from radial glia cells, they have different genomics, genomic landscapes, gene expression, and immunohistochemical signatures.[6,7,8,9] Supratentorial tumors are more often characterized by neuronal differentiation.[7] It is clear that supratentorial and infratentorial ependymomas should be considered separate biological entities.[6,9,10,11,12]
WHO Classification of Tumours Editorial Board, ed.: WHO Classification of Tumours: Central Nervous System Tumours. Vol. 6. 5th ed. IARC Press; 2021.
Louis DN, Ohgaki H, Wiestler OD: WHO Classification of Tumours of the Central Nervous System. 4th rev.ed. IARC Press, 2016.
Pajtler KW, Mack SC, Ramaswamy V, et al.: The current consensus on the clinical management of intracranial ependymoma and its distinct molecular variants. Acta Neuropathol 133 (1): 5-12, 2017.
Louis DN, Perry A, Wesseling P, et al.: The 2021 WHO Classification of Tumors of the Central Nervous System: a summary. Neuro Oncol 23 (8): 1231-1251, 2021.
Ellison DW, Kocak M, Figarella-Branger D, et al.: Histopathological grading of pediatric ependymoma: reproducibility and clinical relevance in European trial cohorts. J Negat Results Biomed 10: 7, 2011.
Taylor MD, Poppleton H, Fuller C, et al.: Radial glia cells are candidate stem cells of ependymoma. Cancer Cell 8 (4): 323-35, 2005.
Andreiuolo F, Puget S, Peyre M, et al.: Neuronal differentiation distinguishes supratentorial and infratentorial childhood ependymomas. Neuro Oncol 12 (11): 1126-34, 2010.
Pajtler KW, Witt H, Sill M, et al.: Molecular Classification of Ependymal Tumors across All CNS Compartments, Histopathological Grades, and Age Groups. Cancer Cell 27 (5): 728-43, 2015.
Johnson RA, Wright KD, Poppleton H, et al.: Cross-species genomics matches driver mutations and cell compartments to model ependymoma. Nature 466 (7306): 632-6, 2010.
Stage Information for Childhood Ependymoma
Although there is no formal staging system, ependymomas are divided into supratentorial, posterior fossa (infratentorial), and spinal tumors. Approximately 20% of childhood ependymomas arise in the spine, and 80% arise in the brain (30% in the supratentorial region and 70% in the posterior fossa).[1]
Ependymomas usually originate in the ependymal linings of ventricles or central canal or ventriculus terminalis of the spinal cord and have access to the cerebrospinal fluid. Therefore, these tumors may spread throughout the neuraxis, although leptomeningeal dissemination is noted in less than 10% of patients with intracranial ependymomas at initial diagnosis.
Myxopapillary ependymoma may disseminate,[2,3] and spinal ependymoma with MYCN amplification shows a high rate of metastasis, with up to 50% of pediatric patients demonstrating leptomeningeal seeding at presentation.[4]
Magnetic resonance imaging of the brain and entire spine, along with lumbar puncture for cytology, is performed at diagnosis to assess for metastatic disease.
References:
Villano JL, Parker CK, Dolecek TA: Descriptive epidemiology of ependymal tumours in the United States. Br J Cancer 108 (11): 2367-71, 2013.
Fassett DR, Pingree J, Kestle JR: The high incidence of tumor dissemination in myxopapillary ependymoma in pediatric patients. Report of five cases and review of the literature. J Neurosurg 102 (1 Suppl): 59-64, 2005.
Bandopadhayay P, Silvera VM, Ciarlini PDSC, et al.: Myxopapillary ependymomas in children: imaging, treatment and outcomes. J Neurooncol 126 (1): 165-174, 2016.
Raffeld M, Abdullaev Z, Pack SD, et al.: High level MYCN amplification and distinct methylation signature define an aggressive subtype of spinal cord ependymoma. Acta Neuropathol Commun 8 (1): 101, 2020.
Treatment Option Overview for Childhood Ependymoma
Many of the improvements in survival in patients with childhood cancer have been made as a result of clinical trials that have attempted to improve on the best available, accepted therapy. Clinical trials in pediatrics are designed to compare new therapy with therapy that is currently accepted as standard. This comparison may be done in a randomized study of two treatment arms or by evaluating a single new treatment and comparing the results with those previously obtained with existing therapy.
Because of the relative rarity of cancer in children, all patients with aggressive brain tumors should be considered for entry into a clinical trial. To determine and implement optimum treatment, review of each case by a multidisciplinary team of cancer specialists who have experience treating childhood brain tumors is required. Radiation therapy for pediatric brain tumors is technically demanding and should be performed in centers that have pediatric experience to ensure optimal results.
Treatment of childhood ependymoma begins with surgery. The type of adjuvant therapy given, such as a second surgery, chemotherapy, or radiation therapy, depends on the following:
Subtype of ependymoma.
Location of the tumor.
Whether the tumor was completely removed during the initial surgery.
Whether the tumor has disseminated throughout the central nervous system.
Child's age.
Table 1 describes the standard treatment options for newly diagnosed and recurrent childhood ependymoma.
Table 1. Standard Treatment Options for Childhood Ependymoma
Dramatic improvements in survival have been achieved for children and adolescents with cancer. Between 1975 and 2020, childhood cancer mortality decreased by more than 50%.[1,2,3] Childhood and adolescent cancer survivors require close monitoring because cancer therapy side effects may persist or develop months or years after treatment. For specific information about the incidence, type, and monitoring of late effects in childhood and adolescent cancer survivors, see Late Effects of Treatment for Childhood Cancer.
References:
Smith MA, Altekruse SF, Adamson PC, et al.: Declining childhood and adolescent cancer mortality. Cancer 120 (16): 2497-506, 2014.
National Cancer Institute: NCCR*Explorer: An interactive website for NCCR cancer statistics. Bethesda, MD: National Cancer Institute. Available online. Last accessed February 25, 2025.
Surveillance Research Program, National Cancer Institute: SEER*Explorer: An interactive website for SEER cancer statistics. Bethesda, MD: National Cancer Institute. Available online. Last accessed December 30, 2024.
Treatment of Childhood Myxopapillary Ependymoma
Myxopapillary ependymoma, considered to be a histological subtype of ependymoma, has a relatively high incidence of central nervous system (CNS) tumor dissemination at diagnosis and at follow-up. Imaging of the complete craniospinal axis at the time of diagnosis and during follow-up is indicated.[1,2] According to the 2021 World Health Organization (WHO) Classification of Tumors of the CNS, myxopapillary ependymoma is now considered WHO grade 2 rather than grade 1 because its recurrence rate is similar to conventional spinal ependymoma and exceeds the rate typical of grade 1 tumors.[3]
Standard treatment options for newly diagnosed childhood myxopapillary ependymoma include the following:
Surgery with or without adjuvant radiation therapy.
Historically, the management of myxopapillary ependymoma consisted of an attempt at en bloc resection of the tumor with no further treatment in the case of a gross-total resection.[4]; [5][Level of evidence C2] However, some practitioners now favor the use of radiation therapy after surgical resection of the primary mass. This practice is based on the finding that dissemination of these tumors to other parts of the neuraxis can occur, particularly after partial resection, and evidence that focal radiation therapy may improve progression-free survival (PFS).[1,4]; [6,7,8][Level of evidence C2]
With the exception of an en bloc gross-total resection where the utility of adjuvant radiation therapy has been debated, radiation therapy is often considered for patients with less than a gross-total resection, a piecemeal resection, or locally recurrent disease after surgery alone. A retrospective single-institution review included 18 pediatric patients with myxopapillary ependymoma.[9]
The study reported poor 5-year and 10-year event-free survival (EFS) rates of 52% and 26%, respectively.
However, these patients had an excellent 10-year overall survival (OS) rate of 100%.
Fifty percent of the patients had metastatic disease at diagnosis and 50% had subtotal resections, but only three patients received radiation therapy (two received focal and one received craniospinal).
The extent of resection did not affect the EFS rate.
Metastatic disease was associated with a worse EFS (10-year EFS rate, 13%), compared with localized disease (57%; P = .07).
This study concluded that despite the high risk of recurrence, patients with myxopapillary ependymoma have an excellent long-term survival. Therefore, radiation therapy should be reserved for patients with symptomatic recurrences to avoid long-term complications from radiation exposure.
However, two reports provided some support for the use of radiation therapy for patients with multifocal spinal myxopapillary ependymoma. The first study included 12 children (aged <21 years) who were treated with limited-volume brain-sparing proton radiation therapy. The median age of patients was 13.5 years. Radiation therapy was given as adjuvant therapy after primary surgery in five patients and for recurrence in seven patients. No patient had previously received radiation therapy. Of the 12 patients, 11 (92%) had evidence of gross disease at the time of radiation therapy, and all but one patient received 54 Gy relative biological effectiveness (RBE) of radiation therapy.[10]
With a median follow-up of 3.6 years (range, 1.8–10.6 years), the 5-year local control rate was 100%, the PFS rate was 92%, and the OS rate was 100%.
One patient developed grade 3 spinal kyphosis after combined surgery and radiation therapy, and one patient developed grade 2 unilateral L5 neuropathy.
A second multi-institutional retrospective study of 60 pediatric and adolescent and young adult (AYA) patients also suggested a benefit of radiation therapy (2000–2020). The median age at radiation therapy was 14.8 years (range, 7.1–26.5 years). The population was high risk because the indications for radiation therapy included gross residual disease, microscopic residual disease, or recurrent or multifocal disease.[11]
At the time of radiation therapy, 45 patients (75.0%) had gross residual disease, and 35 patients (58.3%) had multifocal disease.
Forty-eight patients (80.0%) received involved-field radiation therapy (IFRT), seven (11.7%) received cranial-spinal radiation therapy, and five (8.3%) received whole-spine radiation therapy.
With a median follow-up of 6.2 years (range, 0.6–21.0 years), the 5-year OS rate was 100%, the PFS rate was 60.8%, and the cumulative incidence of local in-field progression rate was 4.1%.
The two local recurrences were in sites of gross residual disease. Of the 18 out-of-field recurrences after radiation therapy, all were superior to the initial treatment field. Nine of these patients experienced intracranial relapse (five of whom had isolated intracranial relapses).
For patients with metastatic myxopapillary ependymoma, there was no significant difference in PFS between patients treated with IFRT (to all sites) and those treated with whole-brain or craniospinal irradiation (P = .283).
On univariate analysis, distant-only recurrence before radiation therapy was significantly associated with shorter time to progression (HR, 4.00; 95% CI, 1.54–10.43; P = .005).
Conclusions from this report include: 1) the risk of recurrence within the radiation field is low, and 2) pediatric and AYA patients with high-risk myxopapillary ependymoma remain at risk for recurrences in the spine above the radiation fields and intracranially after radiation therapy.
References:
Fassett DR, Pingree J, Kestle JR: The high incidence of tumor dissemination in myxopapillary ependymoma in pediatric patients. Report of five cases and review of the literature. J Neurosurg 102 (1 Suppl): 59-64, 2005.
Bagley CA, Kothbauer KF, Wilson S, et al.: Resection of myxopapillary ependymomas in children. J Neurosurg 106 (4 Suppl): 261-7, 2007.
Louis DN, Perry A, Wesseling P, et al.: The 2021 WHO Classification of Tumors of the Central Nervous System: a summary. Neuro Oncol 23 (8): 1231-1251, 2021.
Akyurek S, Chang EL, Yu TK, et al.: Spinal myxopapillary ependymoma outcomes in patients treated with surgery and radiotherapy at M.D. Anderson Cancer Center. J Neurooncol 80 (2): 177-83, 2006.
Bagley CA, Wilson S, Kothbauer KF, et al.: Long term outcomes following surgical resection of myxopapillary ependymomas. Neurosurg Rev 32 (3): 321-34; discussion 334, 2009.
Pica A, Miller R, Villà S, et al.: The results of surgery, with or without radiotherapy, for primary spinal myxopapillary ependymoma: a retrospective study from the rare cancer network. Int J Radiat Oncol Biol Phys 74 (4): 1114-20, 2009.
Agbahiwe HC, Wharam M, Batra S, et al.: Management of pediatric myxopapillary ependymoma: the role of adjuvant radiation. Int J Radiat Oncol Biol Phys 85 (2): 421-7, 2013.
Jeibmann A, Egensperger R, Kuchelmeister K, et al.: Extent of surgical resection but not myxopapillary versus classical histopathological subtype affects prognosis in lumbo-sacral ependymomas. Histopathology 54 (2): 260-2, 2009.
Bandopadhayay P, Silvera VM, Ciarlini PDSC, et al.: Myxopapillary ependymomas in children: imaging, treatment and outcomes. J Neurooncol 126 (1): 165-174, 2016.
Looi WS, Indelicato DJ, Mailhot Vega RB, et al.: Outcomes following limited-volume proton therapy for multifocal spinal myxopapillary ependymoma. Pediatr Blood Cancer 68 (3): e28820, 2021.
Liu KX, Indelicato DJ, Paulino AC, et al.: Multi-institutional Characterization of Outcomes for Pediatric and Young Adult Patients With High-Risk Myxopapillary Ependymoma After Radiation Therapy. Int J Radiat Oncol Biol Phys 117 (5): 1174-1180, 2023.
Treatment of Childhood Nonmyxopapillary Spinal Ependymoma
Standard treatment options for newly diagnosed childhood nonmyxopapillary spinal ependymoma include the following:
Surgery.
Radiation therapy.
Although studies suggest that surgery alone may be adequate for many grade 1 tumors, adjuvant radiation therapy may improve survival in patients with nonmyxopapillary high-grade (2/3) tumors. A bicentric report from the University of Florida and Massachusetts General Hospital supports the use of radiation therapy for tumor control.[1,2,3]
Between 2008 and 2019, 14 pediatric patients with nonmetastatic nonmyxopapillary grade 2 (n = 6) and grade 3 (n = 8) spinal ependymomas were treated with radiation therapy doses between 50.4 Gy relative biological effectiveness (RBE) and 54 Gy RBE (protons). The median age for patients at the time of radiation therapy was 14 years (range, 1.5–18 years). Before radiation therapy, 3 patients underwent subtotal resection, and 11 patients had gross-total or near-total resections.[4]
With a median follow-up of 6.3 years (range, 1.5–14.8 years), no tumors progressed.
Although most patients experienced neurological sequelae after surgery, only one developed additional neurological deficits after radiation therapy.
References:
Oh MC, Ivan ME, Sun MZ, et al.: Adjuvant radiotherapy delays recurrence following subtotal resection of spinal cord ependymomas. Neuro Oncol 15 (2): 208-15, 2013.
Volpp PB, Han K, Kagan AR, et al.: Outcomes in treatment for intradural spinal cord ependymomas. Int J Radiat Oncol Biol Phys 69 (4): 1199-204, 2007.
Typically, all patients undergo surgery to remove the tumor. Whether additional treatment is given depends on the ependymoma subtype, age of the child, extent of tumor resection, and whether disseminated disease is present.
Surgery
Surgery is performed in an attempt at maximal tumor reduction. Evidence suggests that more extensive surgical resection is related to an improved rate of survival.[1,2,3,4,5]; [6,7][Level of evidence C2] Magnetic resonance imaging (MRI) is performed postoperatively to confirm the extent of resection. If not obtained preoperatively, MRI of the entire neuraxis and cerebrospinal fluid cytopathology is performed to evaluate for disease dissemination.
Patients across all molecular subgroups who have residual tumor or disseminated disease are considered at high risk of relapse and may be treated on clinical trials specifically designed for them. Patients with no evidence of residual tumor still have an approximate 20% to 40% relapse risk despite receiving postoperative radiation therapy.[8][Level of evidence B4]
Anecdotal experience suggests that surgery alone for completely resected supratentorial World Health Organization (WHO) grade 2 tumors and spinal ependymomas may, in select cases, be an appropriate approach to treatment.[9,10,11,12,13][Level of evidence C2]
Evidence (surgery):
A prospective multi-institutional cooperative group trial (Children's Oncology Group [COG] ACNS0121 [NCT00027846]) included patients with newly diagnosed intracranial ependymomas (N = 356). Surgery alone was used for the treatment of supratentorial, WHO grade 2, gross-totally resected ependymomas (n = 11).[8][Level of evidence B4]
The 5-year event-free survival (EFS) rate was 61.4%, and the overall survival (OS) rate was 100%.
Local failure occurred in four patients (36%), and local and distant failure occurred in one patient (9%).
In this study, the number of patients eligible for a surgery-alone approach was very small. Only a subset of these patients successfully avoided additional treatment.
Retrospective analysis of the outcome for patients with posterior fossa B ependymoma suggests that these patients might be sufficiently treated with gross-total resection alone,[7] but this approach has not been tested in a prospective randomized clinical trial.
Adjuvant Therapy
Treatment of no residual disease, no disseminated disease
Radiation therapy
The standard postsurgical treatment for these patients has been radiation therapy consisting of 54 Gy to 59.4 Gy to the tumor bed for children aged 3 years and older.[5,14] The ACNS0121 (NCT00027846) study extended the use of radiation therapy (54 Gy) to patients as young as 1 year, resulting in similar EFS and OS rates when compared with children older than 3 years.[8][Level of evidence B4]
It is not necessary to treat the entire CNS (whole brain and spine) because these tumors usually recur initially at the local site, although posterior fossa ependymomas may disseminate at recurrence, particularly in tumors with 1q gain.[15]; [16][Level of evidence C1]
Evidence (radiation therapy):
In one single-institution study, 74 patients aged 1 to 21 years were treated with conformal radiation therapy immediately after surgery.[17]
The 3-year progression-free survival (PFS) rate was 77.6% (± 5.8%).
In an expansion of the above series, 107 of 153 patients received conformal radiation therapy immediately after up-front resection.[5][Level of evidence C1]
The 7-year EFS rate was 76.9% (± 13.5%).
A COG prospective study (ACNS0121 [NCT00027846]) enrolled 356 patients between the ages of 1 and 21 years with newly diagnosed ependymoma into four strata.[8][Level of evidence B4]
Stratum 1: Patients with completely resected differentiated histology supratentorial ependymomas who were treated without radiation therapy.
The 5-year PFS rate was 61.4% (95% confidence interval, 34.5%–89.6%), with no deaths at 7 years, although only 11 patients were enrolled in this stratum.
Stratum 2: Patients with subtotally resected ependymomas (both supratentorial and infratentorial) with more than 5 mm residual disease. Treatment consisted of two cycles of chemotherapy followed by second-look surgery and conformal radiation therapy to the tumor bed (adding a 1-cm target clinical volume). Radiation doses were 54 Gy for patients aged 12 to 18 months and 59.4 Gy for patients older than 18 months.
The 5-year PFS rate was 25% for patients in whom a second surgery was not feasible, and 50% for patients in whom a second surgery resulted in a gross-total resection.
Stratum 3 and stratum 4: Patients with near-total resection (stratum 3) and gross-total resection (stratum 4). Patients aged 12 to 18 months received postoperative radiation therapy doses of 54 Gy, and patients older than 18 months received doses of 59.4 Gy (adding a 1-cm target clinical volume).
The 5-year PFS rate was 68.5% (range, 62.8%–74.2%).
Posterior fossa A ependymoma (PF-EPN-A), 1q balanced (without 1q gain): The 5-year PFS rate was 81.5% (range, 71.5%–91.5%).
PF-EPN-A, 1q gain: The 5-year PFS rate was 35.7% (range, 12.8%–58.6%).
For patients with PF-EPN-A, distant failure was more common in patients with 1q gain than in patients with 1q balanced (without 1q gain).
Supratentorial ependymomas: 30 of 39 patients with supratentorial ependymomas who were tested harbored ZFTA fusions, 23 of whom were in stratums 3 and 4. There was no significant difference in survival. The 5-year OS rates exceeded 80%.
Proton-beam radiation therapy (a type of charged-particle radiation therapy) provides a possible advantage for targeting the tumor (supratentorial or infratentorial) while avoiding critical normal brain and neuroendocrine tissues.
In a report from the Massachusetts General Hospital, 150 patients (aged <22 years) with WHO grade 2 and grade 3 ependymomas were treated with proton radiation therapy between 2001 and 2019. The median follow-up was 6.5 years.[18]
For the intracranial cohort (n = 145), the 7-year EFS rate was 63.4%, the OS rate was 82.6%, and the local control rate was 76.1%.
Fifty-one patients experienced a tumor recurrence: 26 patients (51%) had local failures, 19 patients (37.3%) had distant failures, and 6 patients (11.8%) had synchronous failures.
Of the 150 patients, 116 (77.3%) underwent gross-total resection, 5 (3.3%) underwent near-total resection, and 29 (19.3%) underwent subtotal resection.
For the intracranial cohort, the 7-year EFS rate was 70.3% for patients who underwent a gross-total resection or near-total resection and 35.2% for patients who underwent a subtotal resection.
With multivariate analysis, the effect of tumor excision persisted after controlling for tumor location.
In a combined Massachusetts General Hospital and University of Florida study, 386 children with nonmetastatic intracranial grade 2 and grade 3 ependymomas were treated with proton radiation therapy.[19]
With a median follow-up of 5 years, the 7-year local control rate was 77%, the PFS rate was 63.8%, and the OS rate was 82%.
As with the previous report, subtotal resection was associated with worse local control, PFS, and OS.
Radiation therapy doses of greater than 54 Gy were not associated with improved disease control or survival.
The rate of brain stem toxicity greater than grade 2 was 4%, and two children died of brain stem toxicity.
In the KiProReg study, 105 children with intracranial ependymomas were treated with a median total dose of 59.4 Gy of proton radiation therapy. Children younger than 4 years received 54 Gy. The median follow-up was 1.9 years.[20]
The estimated 3-year OS rate was 93.7%, the local control rate was 74.1%, and the PFS rate was 55.6%.
Multiple surgeries were identified as a risk factor for lower PFS.
There was a low rate of grade 3 toxicities and there were no episodes of symptomatic brain stem necrosis.
Concerns about brain stem toxicity in very young children (aged <3 years) after proton therapy to the posterior fossa have prompted the use of more conservative doses in these children at some centers.[21,22,23]
The International Society of Paediatric Oncology (SIOP) Ependymoma I study included 74 eligible pediatric patients with localized ependymomas. Thirty-three patients underwent a gross-total resection before receiving focal irradiation.[24][Level of evidence B4]
The 5-year EFS rate was 69%, and the 10-year EFS rate was 63%.
The 5-year OS rate was 81%, and the 10-year OS rate was 68%.
Post hoc analysis of known risk factors confirmed the impact of 1q gain, H3K27me3 loss, and hTERT expression.
When possible, pediatric patients should be treated in a center experienced with the delivery of highly conformal radiation therapy (including intensity-modulated radiation therapy or charged-particle radiation therapy [e.g., proton radiation therapy]) to minimize long-term side effects.
Chemotherapy
Current treatment approaches do not include chemotherapy as a standard component of primary therapy for children with newly diagnosed ependymomas that are completely resected. The utility of adjuvant chemotherapy was studied in the completed COG ACNS0831 (NCT01096368) trial. Published results of this trial are forthcoming. There is no evidence that myeloablative chemotherapy [25] improves the outcome for patients with totally resected, nondisseminated ependymomas.
Treatment of residual disease, no disseminated disease
Second-look surgery
Second-look surgery should be considered because patients who have complete resections followed by irradiation have better disease control.[26] In some cases, further surgery can be undertaken after the initial attempted resection if the pediatric neurosurgeon believes that a gross-total resection could be obtained by an alternate surgical approach to the tumor. In other cases, additional up-front surgery is not anticipated to result in a gross-total resection; therefore, adjuvant therapy is initiated with future consideration of second-look surgery.[8]
Radiation therapy
The rationale for radiation therapy, as described in the Treatment of no residual disease, no disseminated disease section above, also pertains to the treatment of children with residual nondisseminated ependymoma. In patients who had a subtotal resection, treatment with radiation therapy results in a 5-year PFS rate of 25%. Outcome is particularly poor for patients with PF-EPN-A,[8] although the outcome may be better for patients with residual tumor within the spinal canal.[27]
Preirradiation chemotherapy
The rationale for using chemotherapy in patients with residual tumor is to attempt to achieve a state of no evidence of disease before the patients undergo radiation therapy, either by achieving a complete response (CR) to chemotherapy alone or by facilitating the likelihood of a gross-total resection at the time of second-look surgery after chemotherapy. The benefit of chemotherapy for residual tumor after up-front surgery is still being investigated.
Evidence (preirradiation chemotherapy with or without surgery):
One study demonstrated a benefit of preirradiation chemotherapy in children with near-total resection (>90% resection), with outcomes similar to those for children achieving a gross-total resection followed by radiation therapy.[28]
The COG ACNS0121 (NCT00027846) trial included two cycles of preirradiation chemotherapy for children with residual disease after up-front surgery (n = 64).[8][Level of evidence B4]
Second-look surgery occurred in 39% of patients (n = 25) (gross-total resection, 56%; near-total resection, 20%; subtotal resection, 24%).
For patients who underwent second-look surgery, the 5-year EFS rate was 50.5%, compared with 28.5% for patients who did not undergo second surgery (P = .12).
A multi-institutional trial for children younger than 3 years used preirradiation chemotherapy, followed by conformal radiation once the child was older than 12 months, followed by maintenance chemotherapy.[29][Level of evidence B4]
Fifty-four patients were enrolled, and 54% of patients (n = 29) underwent a gross-total resection at diagnosis.
Of the remaining 25 patients, 60% (n = 15) underwent a second-look surgery after chemotherapy, with 80% of patients achieving a gross-total resection.
At the time of radiation therapy, 76% of patients had a gross-total resection, 13% of patients had a near-total resection, and 11% of patients had a subtotal resection.
PFS (but not OS) was better for patients who underwent a gross-total resection or near-total resection before radiation therapy than it was for patients who underwent a subtotal resection (4-year PFS rate, 79% for gross-total resection/near-total resection vs. 41.7% for subtotal resection) (P = .024).
The SIOP Ependymoma I study enrolled 74 patients, 41 of whom had a subtotal resection after initial surgical management. The protocol specified that these patients were to receive up to four cycles of preirradiation vincristine, etoposide, and cyclophosphamide (VEC). Of the 41 patients, 10 did not receive the protocol-specified VEC therapy, and 3 patients opted for no further therapy and did not receive radiation therapy.[24][Level of evidence B4]
Of the 29 patients who received VEC, the combined complete response and partial response rate was 65%, which exceeded the prespecified 45% response rate threshold.
Eight of 29 patients had progressive disease at the completion of VEC chemotherapy. The 5-year and 10-year EFS rates were 34%. The 5-year OS rate was 60%, and the 10-year OS rate was 54%.
The study demonstrated chemoresponsiveness in most patients. However, the small number of patients precluded a determination of the added benefit of preirradiation VEC in either facilitating a subsequent gross-total resection or in a survival benefit, compared with patients with a subtotal resection who opted not to receive VEC before radiation therapy.
There is no evidence that high-dose chemotherapy with stem cell rescue is beneficial.[30]; [31][Level of evidence B4]
Treatment of CNS disseminated disease
Radiation therapy
Regardless of the degree of surgical resection, patients with CNS disseminated disease generally receive radiation therapy to the whole brain and spine, along with boosts to local disease and bulk areas of disseminated disease. The traditional local postsurgical radiation doses in these patients are 54 Gy to 55.8 Gy. Doses of approximately 36 Gy to the entire neuraxis (i.e., the whole brain and spine) are also administered but may be modulated depending on the age of the patient.[32] Boosts between 41.4 Gy and 50.4 Gy to bulk areas of spinal disease are administered, with doses depending on the age of the patient and the location of the tumor. However, there are no contemporary studies published to support this approach.
Chemotherapy
While chemotherapy is often used because of some degree of chemoresponsiveness, evidence demonstrating improvement in EFS and OS is lacking.[33]
Treatment of children younger than 1 year
Chemotherapy
Some, but not all, chemotherapy regimens induce objective responses in children younger than 3 years with newly diagnosed ependymomas.[34,35,36,37] The goal of chemotherapy is to avoid radiation, defer radiation until the child is older, or achieve a state of no evidence of disease before undergoing radiation therapy (either by a CR to chemotherapy or by a gross-total resection at time of second-look surgery after chemotherapy). Up to 25% of infants and young children with totally resected disease may achieve long-term survival. These studies have not been molecularly characterized, and it is unclear which patients may benefit from chemotherapy-only regimens. Survivors of chemotherapy-only protocols may eventually receive salvage radiation therapy.[38]; [39][Level of evidence B4]
Deferred radiation therapy
Historically, postoperative radiation therapy was omitted for children younger than 3 years with ependymomas. Two COG studies (POG-9233 and ACNS0121 [NCT00027846]) and many subsequent trials have lowered the age limit for postoperative radiation therapy to 1 year in an effort to improve outcomes for these younger children. The ACNS0121 trial showed that conformal radiation in children with completely resected tumors resulted in significantly improved outcomes compared with patients who received chemotherapy alone.[8][Level of evidence B4]
It is unclear which patients can benefit from radiation-sparing approaches. However, comparison of the POG-9233 trial results with the ACNS0121 (NCT00027846) trial results suggests a 50% to 60% improvement in survival for patients who were treated with radiation therapy.[8,38] A prospective evaluation of molecular markers may identify the infants who can be safely treated with radiation-sparing approaches and/or patients who may benefit from chemotherapy.
Evidence (radiation therapy):
Retrospective reviews based on Surveillance, Epidemiology, and End Results Program data from children younger than 3 years at diagnosis were accrued over a 50-year period.[40]
Results showed that patients who received local radiation therapy had better 10-year survival rates, even after adjusting for the extent of resection and tumor grade (WHO grade 2 vs. grade 3).
A large retrospective study, across 820 molecularly characterized posterior fossa ependymomas, demonstrated the following:[7]
Adjuvant first-line radiation therapy, along with complete resection and PF-EPN-B subgroup, were associated with an improved prognosis.
Radiation-sparing approaches were associated with dismal outcomes in children with PF-EPN-A tumors.
Conformal or charged-particle (e.g., proton) radiation therapy is an alternative approach for minimizing radiation-induced neurological damage in young children with ependymomas. The need and timing of radiation therapy for children who have successfully completed chemotherapy and have no residual disease is still to be determined.
The initial experience with this approach suggested that children younger than 3 years with ependymomas have neurological deficits at diagnosis that improve with time after conformal radiation treatment.[17]
Another study suggested that there was a trend for intellectual deterioration over time, even in older children treated with localized radiation therapy.[41]; [42][Level of evidence C1]
The COG ACNS0121 (NCT00027846) study showed that children aged 1 year to younger than 3 years who underwent a gross-total resection or near-total resection followed by immediate postoperative radiation therapy had the following results:[8][Level of evidence B4]
The 5-year EFS rate was 62.9%, and the OS rate was 87.4%.
These results were not statistically different from the results seen in patients aged 3 to 21 years, who had a 5-year EFS rate of 70.5% and an OS rate of 85.8%.
A multi-institutional trial of children younger than 3 years with newly diagnosed ependymomas (n = 54) who received four to six cycles of chemotherapy followed by radiation therapy (once they had reached the age of 12 months) resulted in the following:[29][Level of evidence B4]
The 4-year PFS rate was 75.1%, and the OS rate was 92.6%.
These results were comparable to the results seen in studies that treated children older than 3 years.
Of interest, there was no difference in outcomes between infants younger than 1 year and children aged 1 to 3 years at diagnosis.
Conformal radiation approaches, including 3-dimensional conformal radiation therapy that minimizes damage to normal brain tissue and charged-particle radiation therapy, such as proton-beam therapy, are under evaluation for infants and children with ependymomas.[17,43] When analyzing neurological outcomes after treatment of young children with ependymomas, it is important to consider that not all long-term deficits can be attributed to radiation therapy, because deficits may be present in young children before therapy begins.[17] For example, the presence of hydrocephalus at diagnosis is associated with a lower intelligence quotient, as measured after surgical resection and before administration of radiation therapy.[44]
Treatment Options Under Clinical Evaluation for Childhood Ependymoma
Early-phase therapeutic trials may be available for selected patients. These trials may be available via the COG, the Pediatric Brain Tumor Consortium, or other entities. Information about National Cancer Institute (NCI)–supported clinical trials can be found on the NCI website. For information about clinical trials sponsored by other organizations, see the ClinicalTrials.gov website.
Current Clinical Trials
Use our advanced clinical trial search to find NCI-supported cancer clinical trials that are now enrolling patients. The search can be narrowed by location of the trial, type of treatment, name of the drug, and other criteria. General information about clinical trials is also available.
References:
van Veelen-Vincent ML, Pierre-Kahn A, Kalifa C, et al.: Ependymoma in childhood: prognostic factors, extent of surgery, and adjuvant therapy. J Neurosurg 97 (4): 827-35, 2002.
Abdel-Wahab M, Etuk B, Palermo J, et al.: Spinal cord gliomas: A multi-institutional retrospective analysis. Int J Radiat Oncol Biol Phys 64 (4): 1060-71, 2006.
Kothbauer KF: Neurosurgical management of intramedullary spinal cord tumors in children. Pediatr Neurosurg 43 (3): 222-35, 2007.
Zacharoulis S, Ji L, Pollack IF, et al.: Metastatic ependymoma: a multi-institutional retrospective analysis of prognostic factors. Pediatr Blood Cancer 50 (2): 231-5, 2008.
Merchant TE, Li C, Xiong X, et al.: Conformal radiotherapy after surgery for paediatric ependymoma: a prospective study. Lancet Oncol 10 (3): 258-66, 2009.
Cage TA, Clark AJ, Aranda D, et al.: A systematic review of treatment outcomes in pediatric patients with intracranial ependymomas. J Neurosurg Pediatr 11 (6): 673-81, 2013.
Ramaswamy V, Hielscher T, Mack SC, et al.: Therapeutic Impact of Cytoreductive Surgery and Irradiation of Posterior Fossa Ependymoma in the Molecular Era: A Retrospective Multicohort Analysis. J Clin Oncol 34 (21): 2468-77, 2016.
Merchant TE, Bendel AE, Sabin ND, et al.: Conformal Radiation Therapy for Pediatric Ependymoma, Chemotherapy for Incompletely Resected Ependymoma, and Observation for Completely Resected, Supratentorial Ependymoma. J Clin Oncol 37 (12): 974-983, 2019.
Volpp PB, Han K, Kagan AR, et al.: Outcomes in treatment for intradural spinal cord ependymomas. Int J Radiat Oncol Biol Phys 69 (4): 1199-204, 2007.
Hukin J, Epstein F, Lefton D, et al.: Treatment of intracranial ependymoma by surgery alone. Pediatr Neurosurg 29 (1): 40-5, 1998.
Little AS, Sheean T, Manoharan R, et al.: The management of completely resected childhood intracranial ependymoma: the argument for observation only. Childs Nerv Syst 25 (3): 281-4, 2009.
Venkatramani R, Dhall G, Patel M, et al.: Supratentorial ependymoma in children: to observe or to treat following gross total resection? Pediatr Blood Cancer 58 (3): 380-3, 2012.
Ghia AJ, Mahajan A, Allen PK, et al.: Supratentorial gross-totally resected non-anaplastic ependymoma: population based patterns of care and outcomes analysis. J Neurooncol 115 (3): 513-20, 2013.
Koshy M, Rich S, Merchant TE, et al.: Post-operative radiation improves survival in children younger than 3 years with intracranial ependymoma. J Neurooncol 105 (3): 583-90, 2011.
Combs SE, Kelter V, Welzel T, et al.: Influence of radiotherapy treatment concept on the outcome of patients with localized ependymomas. Int J Radiat Oncol Biol Phys 71 (4): 972-8, 2008.
Schroeder TM, Chintagumpala M, Okcu MF, et al.: Intensity-modulated radiation therapy in childhood ependymoma. Int J Radiat Oncol Biol Phys 71 (4): 987-93, 2008.
Merchant TE, Mulhern RK, Krasin MJ, et al.: Preliminary results from a phase II trial of conformal radiation therapy and evaluation of radiation-related CNS effects for pediatric patients with localized ependymoma. J Clin Oncol 22 (15): 3156-62, 2004.
Patteson BE, Baliga S, Bajaj BVM, et al.: Clinical outcomes in a large pediatric cohort of patients with ependymoma treated with proton radiotherapy. Neuro Oncol 23 (1): 156-166, 2021.
Indelicato DJ, Ioakeim-Ioannidou M, Bradley JA, et al.: Proton Therapy for Pediatric Ependymoma: Mature Results From a Bicentric Study. Int J Radiat Oncol Biol Phys 110 (3): 815-820, 2021.
Peters S, Merta J, Schmidt L, et al.: Evaluation of dose, volume, and outcome in children with localized, intracranial ependymoma treated with proton therapy within the prospective KiProReg Study. Neuro Oncol 24 (7): 1193-1202, 2022.
Indelicato DJ, Flampouri S, Rotondo RL, et al.: Incidence and dosimetric parameters of pediatric brainstem toxicity following proton therapy. Acta Oncol 53 (10): 1298-304, 2014.
Sato M, Gunther JR, Mahajan A, et al.: Progression-free survival of children with localized ependymoma treated with intensity-modulated radiation therapy or proton-beam radiation therapy. Cancer 123 (13): 2570-2578, 2017.
Indelicato DJ, Bradley JA, Rotondo RL, et al.: Outcomes following proton therapy for pediatric ependymoma. Acta Oncol 57 (5): 644-648, 2018.
Ritzmann TA, Chapman RJ, Kilday JP, et al.: SIOP Ependymoma I: Final results, long-term follow-up, and molecular analysis of the trial cohort-A BIOMECA Consortium Study. Neuro Oncol 24 (6): 936-948, 2022.
Zacharoulis S, Levy A, Chi SN, et al.: Outcome for young children newly diagnosed with ependymoma, treated with intensive induction chemotherapy followed by myeloablative chemotherapy and autologous stem cell rescue. Pediatr Blood Cancer 49 (1): 34-40, 2007.
Massimino M, Solero CL, Garrè ML, et al.: Second-look surgery for ependymoma: the Italian experience. J Neurosurg Pediatr 8 (3): 246-50, 2011.
Wahab SH, Simpson JR, Michalski JM, et al.: Long term outcome with post-operative radiation therapy for spinal canal ependymoma. J Neurooncol 83 (1): 85-9, 2007.
Garvin JH, Selch MT, Holmes E, et al.: Phase II study of pre-irradiation chemotherapy for childhood intracranial ependymoma. Children's Cancer Group protocol 9942: a report from the Children's Oncology Group. Pediatr Blood Cancer 59 (7): 1183-9, 2012.
Upadhyaya SA, Robinson GW, Onar-Thomas A, et al.: Molecular grouping and outcomes of young children with newly diagnosed ependymoma treated on the multi-institutional SJYC07 trial. Neuro Oncol 21 (10): 1319-1330, 2019.
Grill J, Kalifa C, Doz F, et al.: A high-dose busulfan-thiotepa combination followed by autologous bone marrow transplantation in childhood recurrent ependymoma. A phase-II study. Pediatr Neurosurg 25 (1): 7-12, 1996.
Venkatramani R, Ji L, Lasky J, et al.: Outcome of infants and young children with newly diagnosed ependymoma treated on the "Head Start" III prospective clinical trial. J Neurooncol 113 (2): 285-91, 2013.
Merchant TE, Boop FA, Kun LE, et al.: A retrospective study of surgery and reirradiation for recurrent ependymoma. Int J Radiat Oncol Biol Phys 71 (1): 87-97, 2008.
Bouffet E, Capra M, Bartels U: Salvage chemotherapy for metastatic and recurrent ependymoma of childhood. Childs Nerv Syst 25 (10): 1293-301, 2009.
Duffner PK, Horowitz ME, Krischer JP, et al.: The treatment of malignant brain tumors in infants and very young children: an update of the Pediatric Oncology Group experience. Neuro-oncol 1 (2): 152-61, 1999.
Duffner PK, Horowitz ME, Krischer JP, et al.: Postoperative chemotherapy and delayed radiation in children less than three years of age with malignant brain tumors. N Engl J Med 328 (24): 1725-31, 1993.
Geyer JR, Sposto R, Jennings M, et al.: Multiagent chemotherapy and deferred radiotherapy in infants with malignant brain tumors: a report from the Children's Cancer Group. J Clin Oncol 23 (30): 7621-31, 2005.
Grill J, Le Deley MC, Gambarelli D, et al.: Postoperative chemotherapy without irradiation for ependymoma in children under 5 years of age: a multicenter trial of the French Society of Pediatric Oncology. J Clin Oncol 19 (5): 1288-96, 2001.
Strother DR, Lafay-Cousin L, Boyett JM, et al.: Benefit from prolonged dose-intensive chemotherapy for infants with malignant brain tumors is restricted to patients with ependymoma: a report of the Pediatric Oncology Group randomized controlled trial 9233/34. Neuro Oncol 16 (3): 457-65, 2014.
Grundy RG, Wilne SA, Weston CL, et al.: Primary postoperative chemotherapy without radiotherapy for intracranial ependymoma in children: the UKCCSG/SIOP prospective study. Lancet Oncol 8 (8): 696-705, 2007.
Snider CA, Yang K, Mack SC, et al.: Impact of radiation therapy and extent of resection for ependymoma in young children: A population-based study. Pediatr Blood Cancer 65 (3): , 2018.
Zapotocky M, Beera K, Adamski J, et al.: Survival and functional outcomes of molecularly defined childhood posterior fossa ependymoma: Cure at a cost. Cancer 125 (11): 1867-1876, 2019.
von Hoff K, Kieffer V, Habrand JL, et al.: Impairment of intellectual functions after surgery and posterior fossa irradiation in children with ependymoma is related to age and neurologic complications. BMC Cancer 8: 15, 2008.
MacDonald SM, Safai S, Trofimov A, et al.: Proton radiotherapy for childhood ependymoma: initial clinical outcomes and dose comparisons. Int J Radiat Oncol Biol Phys 71 (4): 979-86, 2008.
Merchant TE, Lee H, Zhu J, et al.: The effects of hydrocephalus on intelligence quotient in children with localized infratentorial ependymoma before and after focal radiation therapy. J Neurosurg 101 (2 Suppl): 159-68, 2004.
Treatment of Recurrent Childhood Ependymoma
Recurrence is not uncommon for all grades of ependymoma and may develop many years after initial treatment.[1,2] Late recurrence beyond 10 to 15 years has been reported.[3] Disease generally recurs at the primary tumor site, although concomitant neuraxis dissemination may also be seen. Systemic relapse is extremely rare.
At the time of relapse, a complete evaluation for the extent of recurrence is indicated for all patients.
Treatment options for recurrent childhood ependymoma include the following:
The utility of further surgical intervention is individualized, based on the extent and location of the tumor. A study of 53 patients with recurrent ependymoma demonstrated an improved 5-year overall survival (OS) rate of 48.7% for patients who had gross-total or near-total resections at the time of surgery, compared with 5.3% for patients with less than gross-total or near-total resections.[4][Level of evidence B4]
In some cases, surgically accessible lesions may be treated alternatively with radiation therapy.
Radiation Therapy and/or Chemotherapy
Patients with recurrent ependymomas should be considered for treatment with the following modalities:[5][Level of evidence C1]
Focal retreatment with various radiation modalities, including stereotactic radiosurgery,[6,7,8][Level of evidence C1]; [9,10,11][Level of evidence C2]; [12] intensity-modulated photon therapy, and proton therapy.[13]; [14][Level of evidence C1]
Craniospinal irradiation for both local and distant (spinal) recurrence could be considered.[15] A study of 101 reirradiated patients conducted at St. Jude Children's Research Hospital observed the following results:[13][Level of evidence C2]
The median duration of OS was 75.1 months, and the median freedom from progression was 27.3 months.
The 1-, 2-, and 5-year estimates of OS rates were 95.5%, 74.9%, and 57.3%, respectively.
Among the 46 patients who received focal reirradiation for local failure, 13 had local failures, and 11 had distant-only failures.
Among the ten patients who received craniospinal irradiation for local failure, six had local failures, and none had distant-only failures.
Patients with distant-only failure who were treated with craniospinal irradiation had improved OS compared with individuals with local failure who were treated with focal radiation therapy (hazard ratio [HR], 0.37; 95% confidence interval [CI], 0.16–0.87).
The 10-year cumulative incidence of greater than grade 3 radiation necrosis after repeat radiation therapy was 7.9%.
Gain of chromosome 1q was associated with poorer OS (HR, 3.5; 95% CI, 1.1–10.6) for patients with distant failure (but not local failure) after initial radiation therapy. Other variables associated with reduced OS and freedom from progression included male sex, anaplastic histology at recurrence, and a short interval from initial radiation therapy to recurrence.
In a study of 31 children with locally recurrent ependymomas, patients received local (conformal) radiation at recurrence. They received various fractionation schemes using radiation doses similar to those used at initial diagnosis.[16]
The median local recurrence-free survival was 31 months (range, 2–63 months), and the median OS was 34 months (range, 3–63 months).
Patients who underwent surgery first had significantly higher survival rates than did patients who received reirradiation only.
In contrast, a study of 53 patients with recurrent ependymomas treated with radiation therapy after surgery showed no advantage with the addition of radiation therapy after a gross-total or near-total resection.[4][Level of evidence B4]
There was an improvement in the 5-year OS rate (22% vs. 7%) for patients treated with radiation therapy who had less than gross-total or near-total resections.
Three, and even four, courses of radiation therapy for patients with recurrences can prolong survival with acceptable minimal toxicity.[13][Level of evidence C2]
Active anticancer agents, including cyclophosphamide, cisplatin, carboplatin, lomustine, and etoposide, have been used in the recurrence setting. While older single-agent studies have demonstrated chemoresponsiveness with these agents, response is rarely durable.[17,18]
Regardless of treatment strategy, the prognosis for patients with recurrence is poor.[1] Entry into studies of novel therapeutic approaches should be considered.
Treatment Options Under Clinical Evaluation for Recurrent Childhood Ependymoma
Early-phase therapeutic trials may be available for selected patients. These trials may be available via the Children's Oncology Group (COG), the Pediatric Brain Tumor Consortium, or other entities. Information about National Cancer Institute (NCI)–supported clinical trials can be found on the NCI website. For information about clinical trials sponsored by other organizations, see the ClinicalTrials.gov website.
Current Clinical Trials
Use our advanced clinical trial search to find NCI-supported cancer clinical trials that are now enrolling patients. The search can be narrowed by location of the trial, type of treatment, name of the drug, and other criteria. General information about clinical trials is also available.
References:
Zacharoulis S, Ashley S, Moreno L, et al.: Treatment and outcome of children with relapsed ependymoma: a multi-institutional retrospective analysis. Childs Nerv Syst 26 (7): 905-11, 2010.
Ritzmann TA, Rogers HA, Paine SML, et al.: A retrospective analysis of recurrent pediatric ependymoma reveals extremely poor survival and ineffectiveness of current treatments across central nervous system locations and molecular subgroups. Pediatr Blood Cancer 67 (9): e28426, 2020.
Wu J, Armstrong TS, Gilbert MR: Biology and management of ependymomas. Neuro Oncol 18 (7): 902-13, 2016.
Adolph JE, Fleischhack G, Mikasch R, et al.: Local and systemic therapy of recurrent ependymoma in children and adolescents: short- and long-term results of the E-HIT-REZ 2005 study. Neuro Oncol 23 (6): 1012-1023, 2021.
Messahel B, Ashley S, Saran F, et al.: Relapsed intracranial ependymoma in children in the UK: patterns of relapse, survival and therapeutic outcome. Eur J Cancer 45 (10): 1815-23, 2009.
Kano H, Yang HC, Kondziolka D, et al.: Stereotactic radiosurgery for pediatric recurrent intracranial ependymomas. J Neurosurg Pediatr 6 (5): 417-23, 2010.
Bouffet E, Hawkins CE, Ballourah W, et al.: Survival benefit for pediatric patients with recurrent ependymoma treated with reirradiation. Int J Radiat Oncol Biol Phys 83 (5): 1541-8, 2012.
Desrousseaux J, Claude L, Chaltiel L, et al.: Respective Roles of Surgery, Chemotherapy, and Radiation Therapy for Recurrent Pediatric and Adolescent Ependymoma: A National Multicentric Study. Int J Radiat Oncol Biol Phys 117 (2): 404-415, 2023.
Merchant TE, Boop FA, Kun LE, et al.: A retrospective study of surgery and reirradiation for recurrent ependymoma. Int J Radiat Oncol Biol Phys 71 (1): 87-97, 2008.
Kano H, Niranjan A, Kondziolka D, et al.: Outcome predictors for intracranial ependymoma radiosurgery. Neurosurgery 64 (2): 279-87; discussion 287-8, 2009.
Lin YY, Wu HM, Yang HC, et al.: Repeated gamma knife radiosurgery enables longer tumor control in cases of highly-recurrent intracranial ependymoma. J Neurooncol 148 (2): 363-372, 2020.
Kano H, Su YH, Wu HM, et al.: Stereotactic Radiosurgery for Intracranial Ependymomas: An International Multicenter Study. Neurosurgery 84 (1): 227-234, 2019.
Tsang DS, Burghen E, Klimo P, et al.: Outcomes After Reirradiation for Recurrent Pediatric Intracranial Ependymoma. Int J Radiat Oncol Biol Phys 100 (2): 507-515, 2018.
Eaton BR, Chowdhry V, Weaver K, et al.: Use of proton therapy for re-irradiation in pediatric intracranial ependymoma. Radiother Oncol 116 (2): 301-8, 2015.
Tsang DS, Murray L, Ramaswamy V, et al.: Craniospinal irradiation as part of re-irradiation for children with recurrent intracranial ependymoma. Neuro Oncol 21 (4): 547-557, 2019.
Régnier E, Laprie A, Ducassou A, et al.: Re-irradiation of locally recurrent pediatric intracranial ependymoma: Experience of the French society of children's cancer. Radiother Oncol 132: 1-7, 2019.
Bouffet E, Capra M, Bartels U: Salvage chemotherapy for metastatic and recurrent ependymoma of childhood. Childs Nerv Syst 25 (10): 1293-301, 2009.
Jakacki RI, Foley MA, Horan J, et al.: Single-agent erlotinib versus oral etoposide in patients with recurrent or refractory pediatric ependymoma: a randomized open-label study. J Neurooncol 129 (1): 131-8, 2016.
Latest Updates to This Summary (01 / 06 / 2025)
The PDQ cancer information summaries are reviewed regularly and updated as new information becomes available. This section describes the latest changes made to this summary as of the date above.
Editorial changes were made to this summary.
This summary is written and maintained by the PDQ Pediatric Treatment Editorial Board, which is editorially independent of NCI. The summary reflects an independent review of the literature and does not represent a policy statement of NCI or NIH. More information about summary policies and the role of the PDQ Editorial Boards in maintaining the PDQ summaries can be found on the About This PDQ Summary and PDQ® Cancer Information for Health Professionals pages.
About This PDQ Summary
Purpose of This Summary
This PDQ cancer information summary for health professionals provides comprehensive, peer-reviewed, evidence-based information about the treatment of childhood ependymoma. It is intended as a resource to inform and assist clinicians in the care of their patients. It does not provide formal guidelines or recommendations for making health care decisions.
Reviewers and Updates
This summary is reviewed regularly and updated as necessary by the PDQ Pediatric Treatment Editorial Board, which is editorially independent of the National Cancer Institute (NCI). The summary reflects an independent review of the literature and does not represent a policy statement of NCI or the National Institutes of Health (NIH).
Board members review recently published articles each month to determine whether an article should:
be discussed at a meeting,
be cited with text, or
replace or update an existing article that is already cited.
Changes to the summaries are made through a consensus process in which Board members evaluate the strength of the evidence in the published articles and determine how the article should be included in the summary.
The lead reviewers for Childhood Ependymoma Treatment are:
Kenneth J. Cohen, MD, MBA (Sidney Kimmel Comprehensive Cancer Center at Johns Hopkins Hospital)
Louis S. Constine, MD (James P. Wilmot Cancer Center at University of Rochester Medical Center)
Roger J. Packer, MD (Children's National Hospital)
D. Williams Parsons, MD, PhD (Texas Children's Hospital)
Malcolm A. Smith, MD, PhD (National Cancer Institute)
Any comments or questions about the summary content should be submitted to Cancer.gov through the NCI website's Email Us. Do not contact the individual Board Members with questions or comments about the summaries. Board members will not respond to individual inquiries.
Levels of Evidence
Some of the reference citations in this summary are accompanied by a level-of-evidence designation. These designations are intended to help readers assess the strength of the evidence supporting the use of specific interventions or approaches. The PDQ Pediatric Treatment Editorial Board uses a formal evidence ranking system in developing its level-of-evidence designations.
Permission to Use This Summary
PDQ is a registered trademark. Although the content of PDQ documents can be used freely as text, it cannot be identified as an NCI PDQ cancer information summary unless it is presented in its entirety and is regularly updated. However, an author would be permitted to write a sentence such as "NCI's PDQ cancer information summary about breast cancer prevention states the risks succinctly: [include excerpt from the summary]."
Images in this summary are used with permission of the author(s), artist, and/or publisher for use within the PDQ summaries only. Permission to use images outside the context of PDQ information must be obtained from the owner(s) and cannot be granted by the National Cancer Institute. Information about using the illustrations in this summary, along with many other cancer-related images, is available in Visuals Online, a collection of over 2,000 scientific images.
Disclaimer
Based on the strength of the available evidence, treatment options may be described as either "standard" or "under clinical evaluation." These classifications should not be used as a basis for insurance reimbursement determinations. More information on insurance coverage is available on Cancer.gov on the Managing Cancer Care page.
Contact Us
More information about contacting us or receiving help with the Cancer.gov website can be found on our Contact Us for Help page. Questions can also be submitted to Cancer.gov through the website's Email Us.
This information does not replace the advice of a doctor. Ignite Healthwise, LLC disclaims any warranty or liability for your use of this information. Your use of this information means that you agree to the Terms of Use and Privacy Policy. Learn how we develop our content.
Healthwise, Healthwise for every health decision, and the Healthwise logo are trademarks of Ignite Healthwise, LLC.
The Health Encyclopedia contains general health information. Not all treatments or services described are covered benefits for Kaiser Permanente members or offered as services by Kaiser Permanente. For a list of covered benefits, please refer to your Evidence of Coverage or Summary Plan Description. For recommended treatments, please consult with your health care provider.